ABSTRACT
- The increase of sequence data in public nucleotide databases has made DNA sequence-based identification an indispensable tool for fungal identification. However, the large proportion of mislabeled sequence data in public databases leads to frequent misidentifications. Inaccurate identification is causing severe problems, especially for industrial and clinical fungi, and edible mushrooms. Existing species identification pipelines require separate validation of a dataset obtained from public databases containing mislabeled taxonomic identifications. To address this issue, we developed FunVIP, a fully automated phylogeny-based fungal validation and identification pipeline (https://github.com/Changwanseo/FunVIP). FunVIP employs phylogeny-based identification with validation, where the result is achievable only with a query, database, and a single command. FunVIP command comprises nine steps within a workflow: input management, sequence-set organization, alignment, trimming, concatenation, model selection, tree inference, tree interpretation, and report generation. Users may acquire identification results, phylogenetic tree evidence, and reports of conflicts and issues detected in multiple checkpoints during the analysis. The conflicting sample validation performance of FunVIP was demonstrated by re-iterating the manual revision of a fungal genus with a database with mislabeled sequences, Fuscoporia. We also compared the identification performance of FunVIP with BLAST and q2-feature-classifier with two mass double-revised fungal datasets, Sanghuangporus and Aspergillus section Terrei. Therefore, with its automatic validation ability and high identification performance, FunVIP proves to be a highly promising tool for achieving easy and accurate fungal identification.
-
Keywords: automatic pipeline, DNA barcoding, fungal identification, mislabeled sequences, sequence validation, tree interpretation
Introduction
The kingdom Fungi encompasses an estimated 2.2–3.8 million (Hawksworth and Lücking, 2017) or up to 12 million species (Bhunjun et al., 2022), of which 161,455 species names have been currently accepted (www.speciesfungorum.org/Names/Names.asp, accessed in 2024. 10. 07). Fungi exhibit various trophic modes (such as saprotrophs, endophytes, and pathogens) and growth forms (including mycelia and yeasts) across diverse environments (Alexopoulos et al., 1996; Nguyen et al., 2016; Tedersoo et al., 2014). Fungi have evolved to adapt to diverse environments by producing various enzymes and metabolites. These characteristics have facilitated the study of fungi in various fields of research, such as agriculture, ecology, pathology, and pharmacology (Köhler et al., 2017; Nilsson et al., 2019a). Misidentification of fungal species is causing inappropriate application in the industry (Lücking et al., 2021; Tomé et al., 2019) and incorrect diagnosis and treatment in health care (Spivak and Hanson, 2018) and forms a threat to food safety and public health (Frisvad et al., 2006; Wei et al., 2022). Thus, accurate species identification is required for scientific communication in and across these fields. Traditional fungal identification relied on morphological and phenotypic characteristics (Hawksworth et al., 1996; Kandawatte et al., 2021), but complex growth morphologies, sexual dimorphism, convergent evolution, and phenotypic plasticity have made precise fungal identification difficult (Atkins and Clark, 2004; Hibbett and Taylor, 2013; Taylor, 2011).
To address these challenges, the reliance on molecular identification methods has increased in fungal systematics since the early 1990s (Hebert et al., 2003; Hibbett, 1992; White et al., 1990). Compared to morphology, DNA sequences offer more independent characters, leading to a more robust estimates of fungal phylogeny (Hillis, 1987). In 2012, the internal transcribed spacer (ITS) was designated as a universal barcode marker for the kingdom Fungi (Schoch et al., 2012). Since then, a standardized sequencing protocol for the barcode sequence has led to a substantial accumulation of DNA sequence data in global public databases, such as GenBank (Sayers et al., 2023) and UNITE (Nilsson et al., 2019b). The increase of sequence data in public nucleotide databases has made DNA sequence-based identification an indispensable tool for fungal species identification (Chase and Fay, 2009; Hibbett et al., 2016). The Basic Local Alignment Search Tool (BLAST) (Camacho et al., 2009) is a widely utilized method for sequence-based identification, recognized for its standardization and accessibility (Kõljalg et al., 2005; Sokoł et al., 1999). Using an accurate database is crucial for achieving reliable BLAST results (Hawksworth, 2004; Meiklejohn et al., 2019; Somervuo et al., 2016). However, a large proportion of data in public databases is inaccurate (up to 30%) (Hofstetter et al., 2019; Lücking et al., 2020a; Nilsson et al., 2006) causing widespread and frequent misidentifications in fungal identifications (Tedersoo et al., 2022).
Phylogeny-based identification offers a solution to prevent misidentifications caused by databases with mislabeled sequences. Even so, issues like inappropriate genetic marker selection can mislead the user (Lücking and Hawksworth, 2018). Extensive taxonomic knowledge and the ability to evaluate a phylogenetic tree are required to recognize faulty data and accurately identify a species. Moreover, the lack of standardization in phylogeny-based identification may result in different outcomes depending on the user, even with the same input file. This reduces the productivity, scalability, and reproducibility of phylogeny-based identification (Raja et al., 2017). Therefore, to achieve a uniform and accurate result of phylogeny-based identification, objective decisions should be made in steps such as database collection, outgroup selection, trimming, alignment editing, and phylogenetic tree interpretation. Developing a standardized phylogeny-based identification method would improve accuracy and reduce reliance on expertise.
Efforts to develop an automated phylogenetic pipeline aimed to standardize the species identification processes. The automated phylogenetic pipeline applicable for species identification began with Phylogeny.fr (Dereeper et al., 2008) and recently advanced to One-click Fungal Phylogenetic Tool (OFPT) (Zeng et al., 2023). However, these tools require users to interpret the phylogenetic tree outcome for species identification, thus not being fully automated. Fully automated phylogenetic pipelines, such as SUPERSMART (Antonelli et al., 2017) and OneTwoTree (Drori et al., 2018) were designed to examine the evolutionary relationship among well-established species. However, these tools are impractical for identification purposes. Regarding mislabeled data, the Semi-Automatic Taxonomic Improvement and Validation Algorithm (SATIVA) (Kozlov et al., 2016) validates conflicting phylogenetic trees but has not been integrated into an automatic identification pipeline. Thus, at present, there is no rapid, practical tool available for identifying fungal species in a phylogenetic context (Table 1).
To address the current challenges in fungal identification, we developed FunVIP, an automated phylogeny-based validation and identification pipeline for fungi. This pipeline only requires one table-styled query file from the user, one table-style file of database references, and a single command line. The user can retrieve data at any checkpoint (e.g., alignment, trimming, and the raw tree inference step) and inspect the result. In this paper, we demonstrate the validation ability and identification accuracy of FunVIP through three dataset examples: Fuscoporia (Basidiomycota) dataset (Cho et al., 2023), Sanghuangporus (Basidiomycota) dataset (Shen et al., 2021), and Aspergillus section Terrei (Ascomycota) dataset (Wu et al., 2022). These datasets contain numerous misidentifications but have recently undergone manual re-identification and revision based on phylogenetic analysis by taxonomic experts. We present the validation performance of FunVIP through analysis of the original Fuscoporia dataset to evaluate the ability of FunVIP to recognize mislabeled samples. Additionally, we demonstrate the identification performance of FunVIP by analyzing the original Sanghuangporus and Aspergillus section Terrei datasets. We compare the results with BLAST and q2-feature-classifier to highlight the superior identification accuracy of FunVIP. According to our findings, FunVIP facilitates user-friendly fungal identification based on phylogenetic analysis while minimizing errors.
Materials and Methods
Workflow
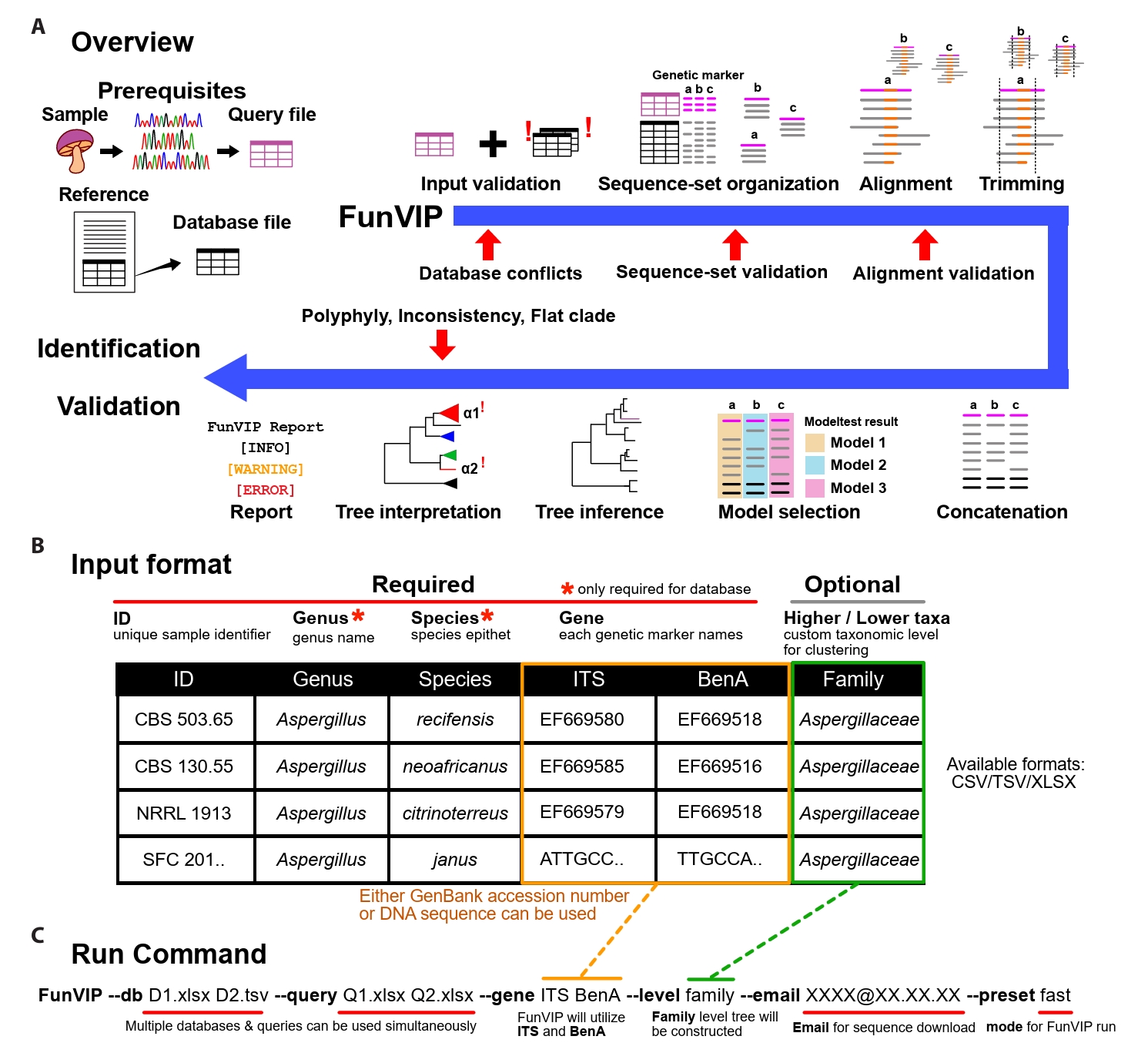
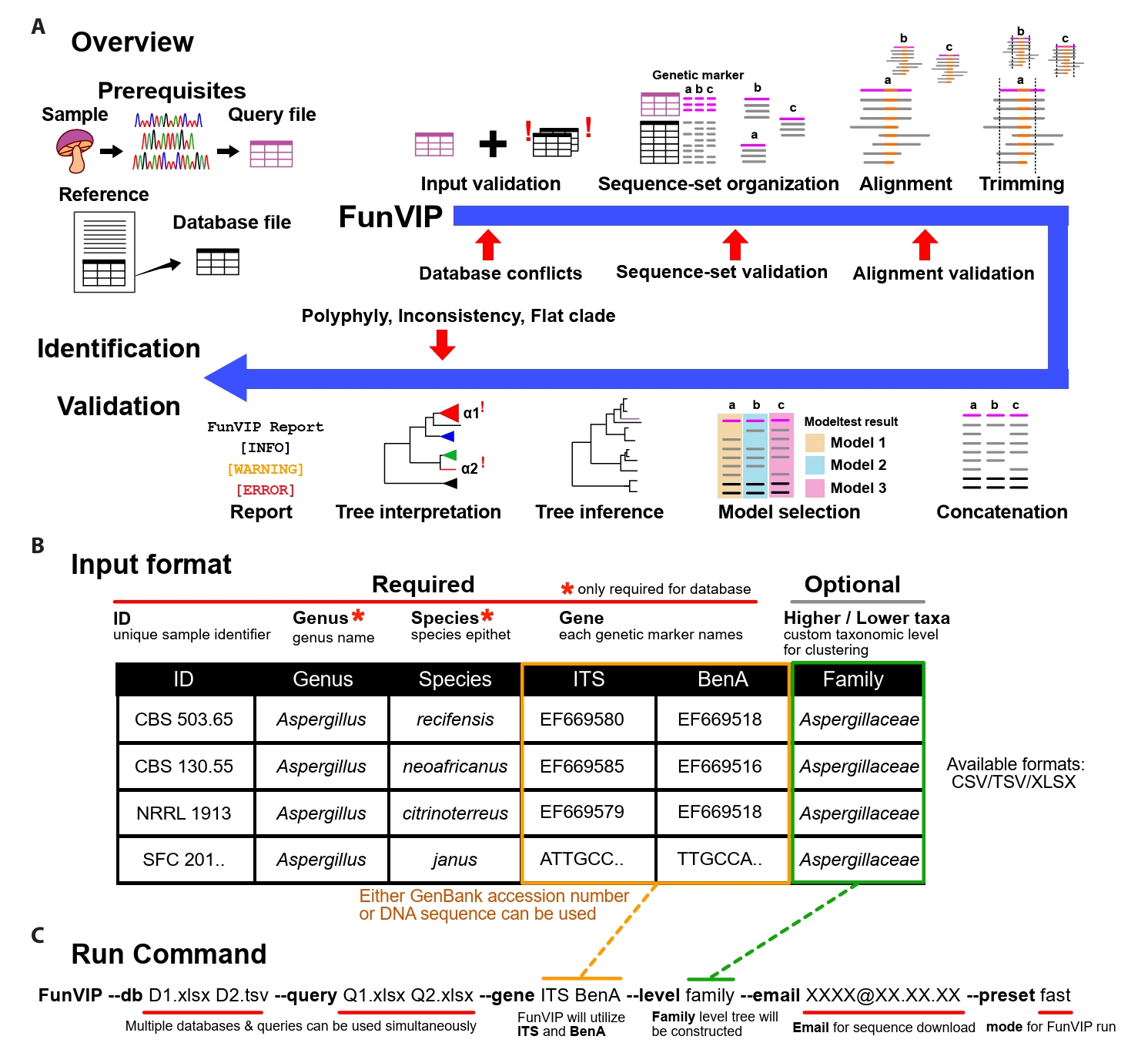
Implementation of FunVIP: FunVIP is an automated pipeline for fungal identification and validation based on phylogenetic analysis. Within a single execution of FunVIP, nine steps are included: input validation, dataset generation, alignment, trimming, concatenation, model selection, tree inference, tree interpretation, and report generation (Fig. 1A). Single node hardware with at least 8 GB is required to run FunVIP, depending on the size of the dataset and the number of threads to be utilized. The pipeline is mostly written in Python 3, and it is compatible with Windows, Mac (Apple Silicon), and Linux platforms. A Conda environment is required to install and execute FunVIP.
Prerequisites: FunVIP requires a table of query files, a table of database files, and a single command line string as input (Fig. 1A). The query table contains unique sample IDs (identifiers indicating biological individual, e.g., strain number or specimen number) and sequences for each genetic marker (GenBank accession numbers or nucleotide sequences). The genus name and species epithet columns can optionally be applied to the query for additional comparison of FunVIP-based identification with the given taxonomy annotation. The database table contains sample IDs, sequences of each gene name, genus name, species epithet, and optional metadata (Fig. 1B). The run command consists of the designation of query and database table files and applicable options, such as mode (fast or accurate) (Fig. 1C). For advanced usage, users can select options for each step from a list of possible choices, with the first option set as the default option (Supple Data 1). Detailed instructions and tips for preparing database and query files for FunVIP is available on tutorial page (https://github.com/Changwanseo/FunVIP/blob/main/tutorial/tutorial2.md).
Input validation step: FunVIP performs input validation to detect and curate human errors before taxonomic analysis. Specifically, FunVIP detects and curates invalid characters in IDs, conflicting sample information, invalid sequence format, and mislabeled genetic markers for both database and query files. For the ID curation, FunVIP automatically corrects characters causing errors during pipeline execution (e.g., umlauts, Latin small ligature “fi”, non-breaking spaces, and whitespaces). In cases of conflicting sample information, where identical sample IDs are associated with different sequences or genetic markers, FunVIP reports the conflicts to guide users in resolving them. For sequence format validation, FunVIP determines whether the sequence is provided as a GenBank accession, DNA string, or FASTA format (Pearson and Lipman, 1988). If a GenBank accession is provided, FunVIP checks whether the accession is valid and retrieves the corresponding sequence using GenMine (Seo et al., 2022). When sequences are provided as raw DNA strings, FunVIP checks for invalid DNA bases. When genetic marker is not given, FunVIP uses BLAST or MMseqs2 (Steinegger and Söding, 2017) to assign the appropriate genetic marker, and excludes invalid data from further analysis.
Sequence-set organization step: Many fungal barcodes cannot be reliably aligned over certain taxonomic levels (Anslan et al., 2018; Tedersoo et al., 2022), dataset division by taxonomic level is required for accurate phylogenetic analysis (Supple Data 2.1, Fig. S2.1). Also, topological incongruences across genetic marker trees should be compared to find out potential mislabels and data contamination in genetic marker annotation. In the sequence-set organization step, FunVIP reorganizes given database and query sequences by taxonomic group-genetic marker combinations to address these needs (Supple Data 2.1, Fig. S2.1).
FunVIP performs sequence-set organization with the consideration that the given database may include mislabeled sequences. For each sequence-set, ingroup, suspicious and outgroup sequences are collected from databases (Supple Data 2.3, Fig. S2.3). Suspicious sequences refer to potential ingroup sequences that are suspected to be misannotated as other taxa (Supple Data 2.4, Fig. S2.4). Outgroup sequences, validated to be outside the target taxonomic group, are sorted into the sequence-set (Supple Data 2.5, Fig. S2.5). Query sequences are sorted into the appropriate sequence-set based on their closest taxonomic assignment. The performance of the query sequence assignment was validated with UFCG dataset (Supple Data 2.6, Fig. S2.6). All sequence distance calculations required for assigning sequences to sequence-sets are determined by pairwise BLAST or MMseqs match. Checkpoints throughout the sequence-set organization step report warnings for users if any issue arises or if the database is insufficient.
Multiple sequence alignment, trimming, concatenation, model selection, and tree inference steps: FunVIP aligns each sequence-set using MAFFT version 7 (Katoh and Standley, 2013) with --maxiterate 1000 option for iterative refinement. Reverse complementary sequences are automatically redirected using --adjustdirection option of MAFFT. The alignments are trimmed using trimAl (Capella-Gutiérrez et al., 2009) or Gblocks (Castresana, 2000). To maximize the number of informative characters, complementary scripts that prevent trimming non-terminal columns of the alignments are included (Supple Data 3.1, Fig. S3.1). A checkpoint in the trimming step assesses whether the alignment meets the criteria necessary infer a valid phylogenetic tree, requiring at least four sequences and at least one fully aligned column across all the alignments (Supple Data 3.2, Fig. S3.2). The Transitive Consistency Score (TCS) (Chang et al., 2014) is calculated with T-Coffee (Notredame et al., 2000) to filter out misaligned sequences. Concatenated multiple genetic marker matrices are generated for each taxonomic group. The nucleotide substitution model for partitions is selected and applied through ModelTest-NG (Darriba et al., 2020) or ModelFinder (Kalyaanamoorthy et al., 2017). Phylogenetic trees are inferred from both individual genetic markers and concatenated matrices using FastTree 2 (Price et al., 2010), IQ-TREE 2 (Minh et al., 2020), or RAxML version 8 (Stamatakis, 2014).
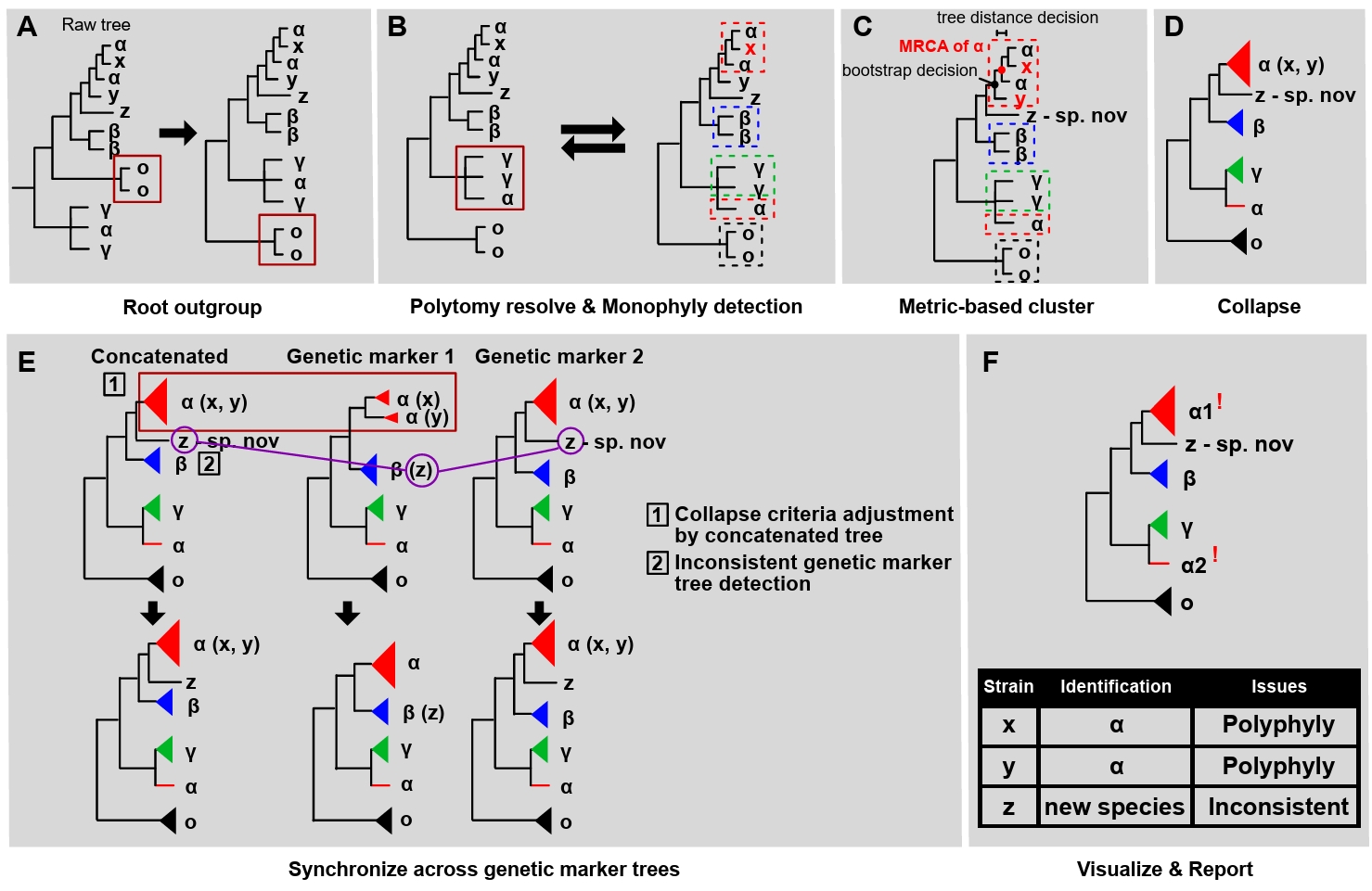
Tree interpretation step: The tree interpretation step involves rooting of the outgroups, polytomy (non-bifurcating branches) resolving, monophyly detection, metric-based clustering, collapsing, synchronizing, visualizing, and reporting (Fig. 2, Supple Data 4, Fig. S4.1). The tree interpretation step deals with three kinds of potential issues: flat branches (Fig. 2B), polyphylies, and inconsistencies (Fig. 2F). Flat branches (polytomies with zero-length branches) are inspected to determine whether the genetic marker has sufficient resolution to resolve taxa at the species level. Polyphylies (multiple monophyletic clades annotated under an identical taxon name) are numbered to inform users to check for misidentified species in the database. Inconsistencies (identification obtained from each of the genetic marker trees differing from the concatenated tree) are inspected to check for mislabeled genes. FunVIP generates a report in table format for the identification result and all potential issues detected during the pipeline run, allowing users to recognize and take appropriate action. It also visualizes phylogenetic trees in a vector image format suitable for further editing.
Performance evaluation
All processes in performance evaluation section were performed with Ryzen 3900X (12 cores, 24 threads) processor and 64 GB memory with Ubuntu 20.04 environment.
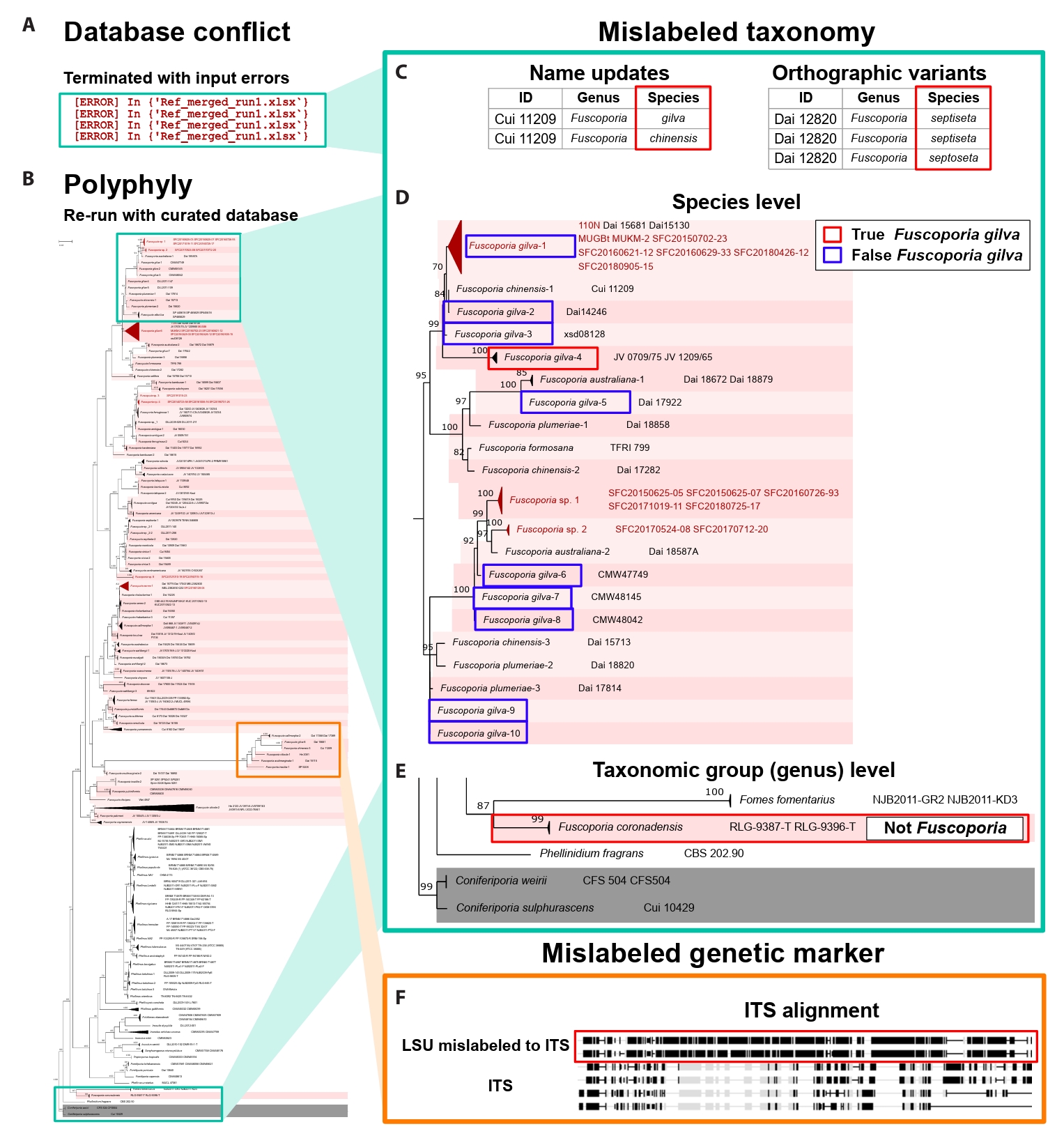
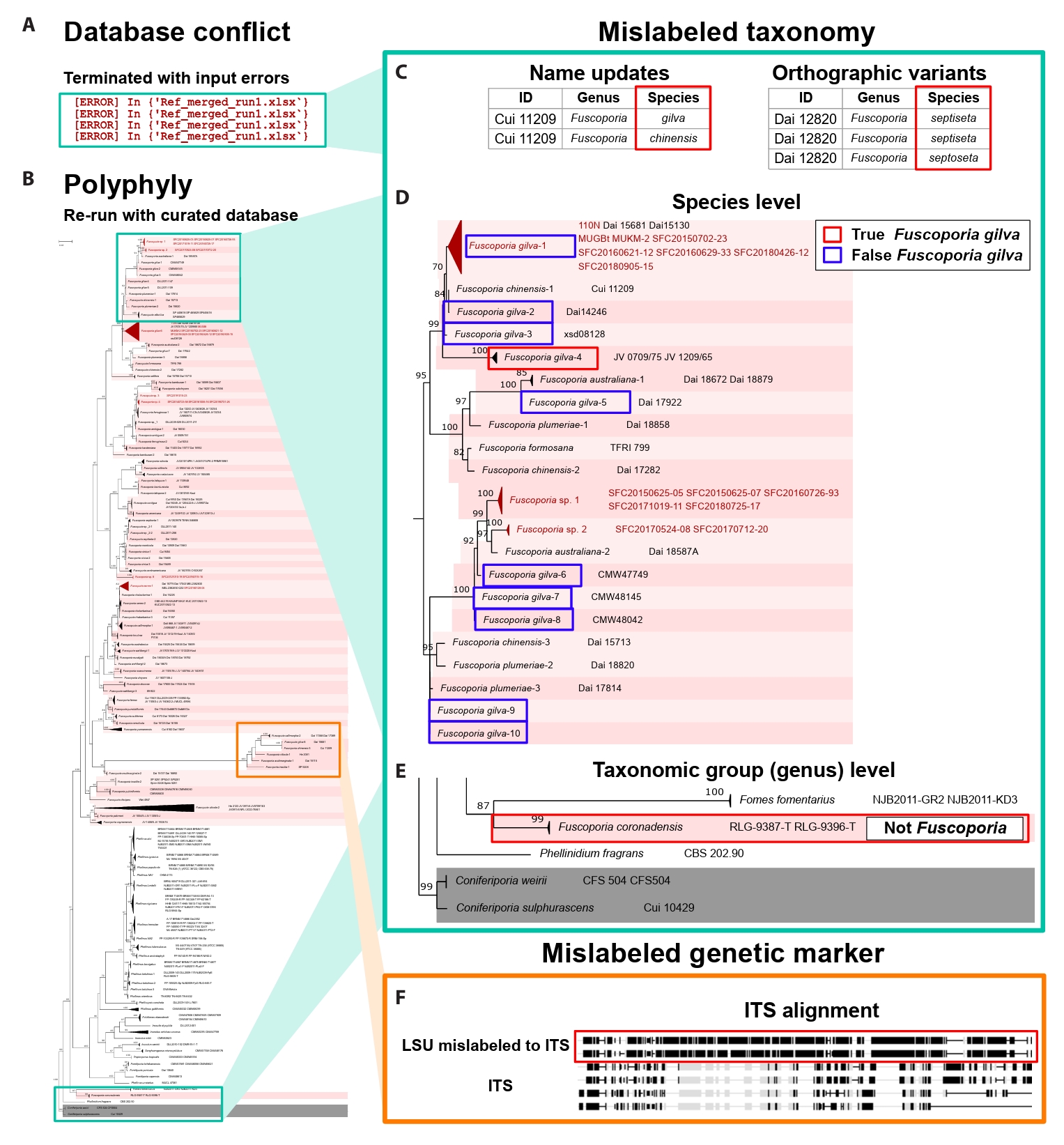
Database validation performance of FunVIP: Validation ability is defined by finding misannotated sequences within the given dataset. To demonstrate the database validation ability of FunVIP, the Fuscoporia dataset was selected as a case study, as it has numerous incorrect identifications, making it an ideal example to illustrate these issues. Taxonomic data tables from seven references indicated in Table 1 of Cho et al. (2023) were prepared. All tables were reformatted to be suitable for valid FunVIP input. The database tables from the seven references were merged into one file for better manipulation. Detailed processes held to prepare input for the analyses are described in Supple Data 5.1.
The first run of FunVIP was performed with the database file and query file in fast mode. Database conflicts (incongruent genus, species, or sequence information among database samples with identical IDs) were found during the input validation step and recorded to the log file (Fig. 3A). The database file was curated according to the conflict report from the first run. For every conflict, the decision of the original reference was applied. The second run was performed with the curated database file and query file in fast mode, and polyphylies found in the concatenated tree were counted from the “ISSUES” column of the report file (Fig. 3B). Detailed processes held during the analyses are described in Supple Data 5.2.
Identification performance of FunVIP: To assess the performance of FunVIP in species identification, we compared FunVIP with the results of BLAST searches (Camacho et al., 2009) and the q2-feature-classifier (Bokulich et al., 2018) using two fungal sequence datasets: Sanghuangporus in Basidiomycota (Shen et al., 2021) and Aspergillus section Terrei in Ascomycota (Wu et al., 2022). These datasets represent sequences of fungal taxa that have been re-identified and revised by taxonomic experts based on phylogenetic analysis. We selected these datasets for evaluating identification performance because expert-revised taxonomic annotation is essential for an accurate identification performance assessment.
The Sanghuangporus dataset comprised ITS sequences of 271 samples, including 15 database and 256 query samples collected from the study by Shen et al. (2021). Two Tropicoporus sequences were included as outgroup samples (Wu et al., 2022). The Aspergillus section Terrei dataset comprised data from four genetic markers [ITS, β-tubulin (BenA), calmodulin (CaM), and RNA polymerase II (RPB2)], including 56 database (outgroup included) and 79 query samples collected from the study by Barros et al. (2020). Manual correction of both datasets, such as orthographic variants and mislabels were performed (Supple Data 6.1, 6.2).
To compare identification performance, multiple options were used to evaluate consistency in each analysis (Supple Data 6.3). A combination of --preset (fast or accurate) and --collapsedistcutoff (0.005–0.3) options were used for FunVIP. The local version of BLAST was performed with database and query files containing the same contents to control variables. Species of the BLAST match with the highest percent identity and within the selected percent identity cutoff (99.5–70) were selected as the identification result. The taxonomic assignment results of q2-feature-classifier were directly applied for the identification result. Accuracy, precision, recall, and F1 score were utilized as statistics to evaluate the performance of identification. Due to the limitation of BLAST and q2-feature-classifier, delimitation of individual new species (e.g., sp. 1 and sp. 2) is not accounted for in the statistics (all regarded as “sp.”). The Mann-Whitney U test was performed for each pair of mean and standard deviation of accuracies to assess the statistical significance.
Results
Validation performance of FunVIP
A total of 501 database samples and 22 query samples were applied in the first run of FunVIP with the Fuscoporia dataset. A total of 113 database conflicts were found during the first run, including ten errors and 103 minor conflicts. The ten errors were caused by six name updates in the subsequent reference (i.e. F. wahlbergii re-identified as F. australasica) and four orthographic variants (i.e. F. setifer as a typo of F. setifera) (Fig. 3C). The other 103 minor conflicts found in the database included two cases where partial sequences with identical sample ID but different accessions (i.e. JQ794579 and JQ794580 both uploaded as the ITS sequence of sample JV0610/14PK). The remaining 101 cases involved sequences that were differentially present among the same sample ID [i.e., the TEF1 sequence is present only for the Cui 11801 sample in the latter reference (Du et al., 2020) and is absent in prior reference (Chen and Dai, 2019)].
The number of samples in the database decreased from 501 to 356 after applying curation according to the conflicts found in the first run and removing duplicates. Among the 356 database samples, 108 samples representing polyphyly were found from the concatenated tree, comprising 20 species and 50 clades. Among the 108 polyphyly samples, 100 samples (44 clades) were caused by mislabeled taxonomies (i.e., F. gilva divided into 10 different clades, with only clade four remaining as the actual F. gilva species) (Fig. 3D). Two samples from one species (annotated as F. coronadensis) turned out to be non-Fuscoporia (Fig. 3E). The remaining seven samples (six clades) were revealed to have mislabeled genetic marker issues, where LSU sequences were labeled as ITS sequences (Fig. 3F).
Identification performance evaluation
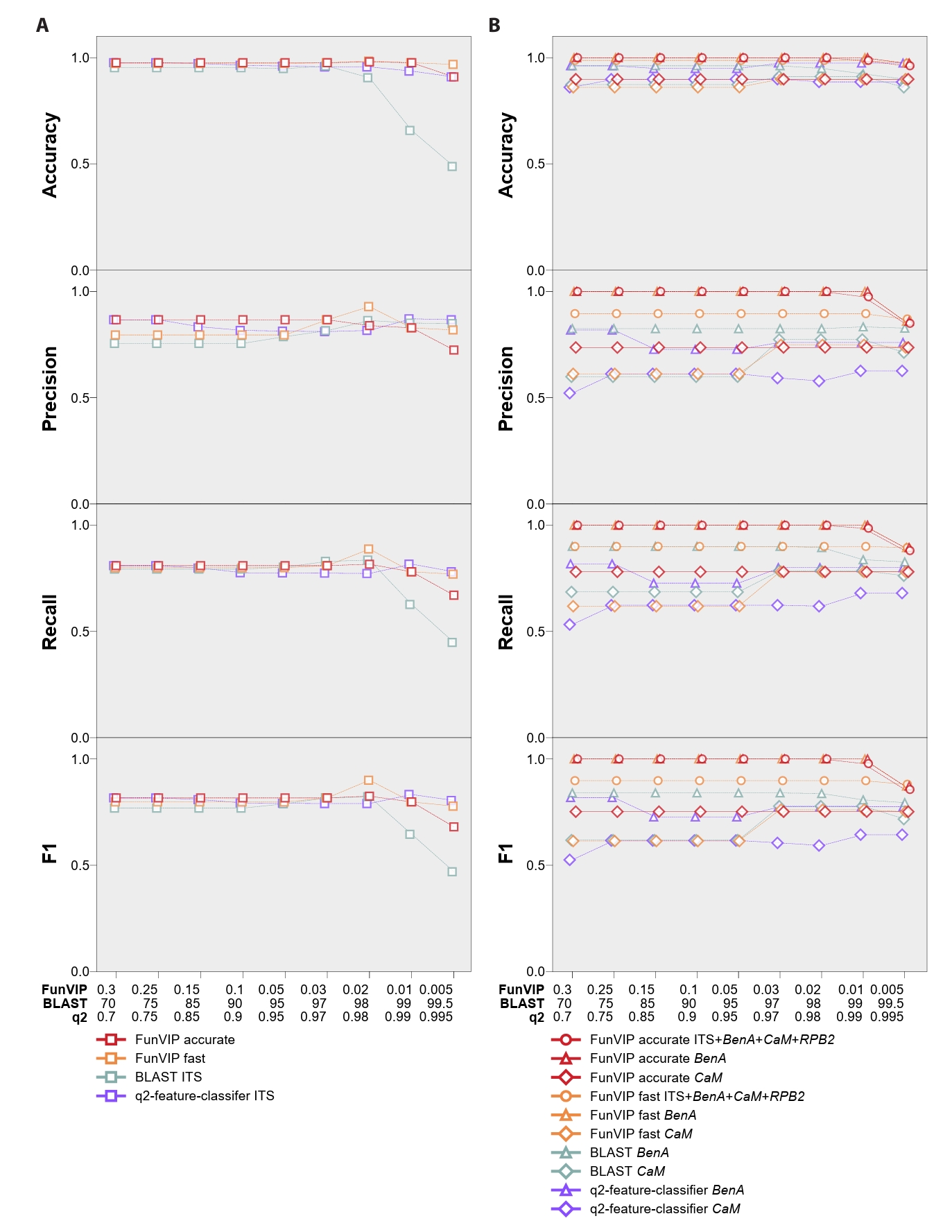
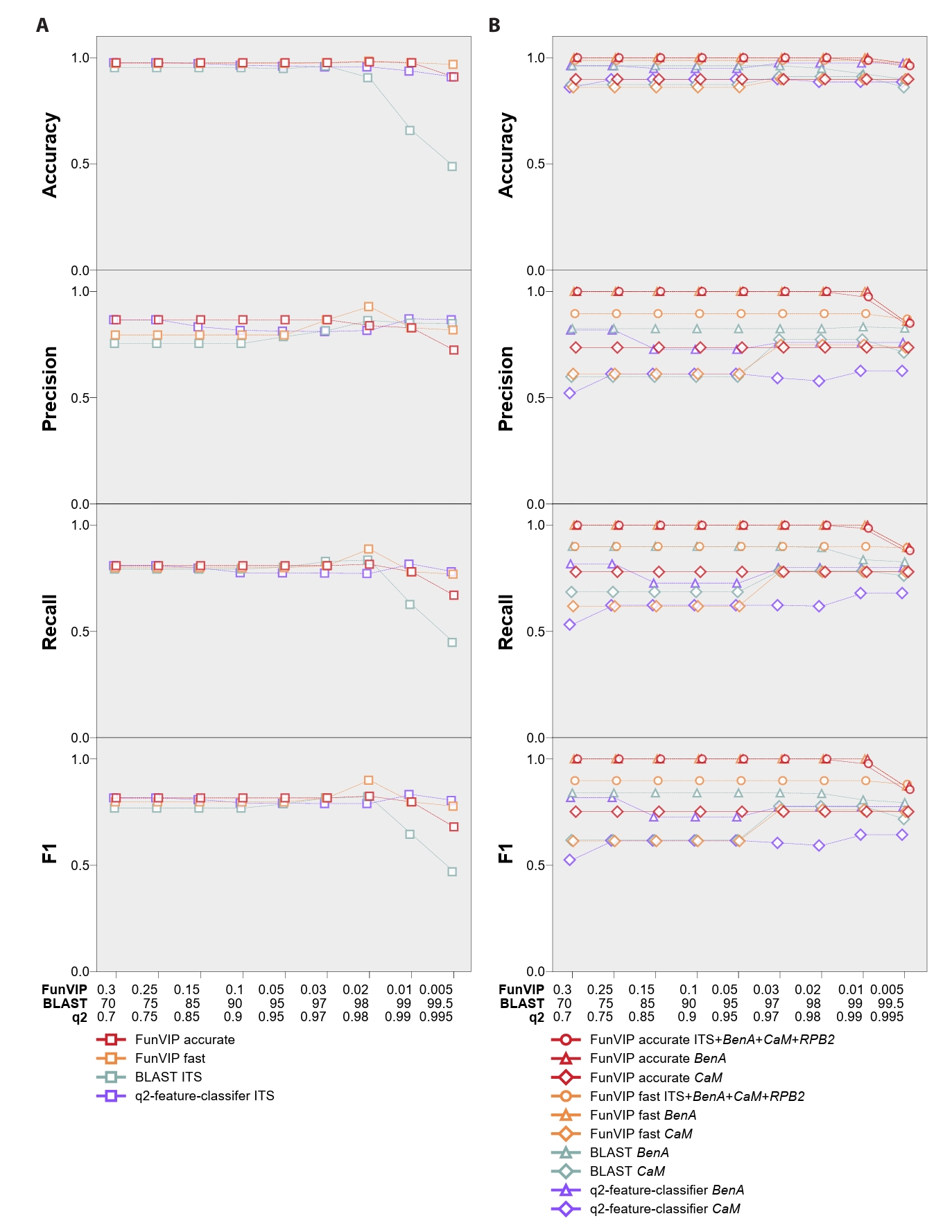
FunVIP exhibited better performance across the majority of the four metrics assessed compared to BLAST and q2-feature-classifier (Fig. 4, Supple Data 7, Table S7.1–7.4). In the Sanghuangporus dataset, the mean identification accuracies were 94.25% in the fast mode and 97.28% in the accurate mode, both of which significantly surpassed the 87.24% performance of BLAST and slightly better than the 95.67% performance of q2-feature-classifier (Fig. 4A). When using the default option of FunVIP (--collapsedistcutoff 0.01) in the accurate mode, six out of 254 entries (2.24%) were misidentified due to systematic underclassification [Sanghuangporus sp. 3 misidentified as S. microcystideus (n=4)] and overclassification [S. weirianus identified as Sanghuangporus sp. 2 (n=1), S. zonatus identified as Sanghuangporus sp. 4 (n=1)]. The standard deviation of accuracy was lower for FunVIP (fast: 0.0042, accurate: 0.0212) than for BLAST (0.1552), and similar to those of q2-feature-classifier (0.0203) indicating that FunVIP is more consistent than BLAST.
For the Aspergillus section Terrei dataset, FunVIP achieved mean identification accuracies of 98.6% in the fast mode and 99.4% in the accurate mode, significantly outperforming BLAST with BenA (95.1%) and with CaM (88.4%) (Fig. 4B). FunVIP also achieved significantly better performance than q2-feature-classifier with BenA (94.9%) and CaM (88.5%). Moreover, the standard deviations of accuracy across multiple options (0.0040 in fast mode, 0.0121 in accurate mode) were significantly lower for FunVIP than for BLAST (BenA: 0.0215, CaM: 0.0193), and similar to those of q2-feature-classifier (0.0111, 0.0119). In the default option of FunVIP (--collapsedistcutoff 0.01) in the accurate mode, only one sample, URM3371 A. recifensis was misidentified to Aspergillus sp. 1.
Discussion
The demands for precise fungal identification are increasing across diverse scientific fields, including taxonomy, medical and nutritional sciences, clinical practice, and quarantine management. However, consistent generation of mislabeled sequences poses a significant obstacle to accurate sequence-based identification. To address this challenge, we developed FunVIP to provide a user-friendly and intuitive solution for species identification.
FunVIP: a superior species identification pipeline based on phylogeny
Practical automated phylogeny-based pipelines fungal identification pipelines are scarce due to challenges in standardization and expertise requirements. A standardized phylogenetic analysis is important for objective identification and cross-validation (Dissanayake et al., 2020). However, variables such as alignment editing, trimming, and branch length criteria for species delimitation are often subjective, which can lead to irreproducible and incoherent phylogenetic trees (Petersen et al., 2010). FunVIP addresses these challenges by allowing users to define ambiguous variables digitally and apply advanced options that can be saved as preset files for reproducibility (Supple Data 1). For instance, alignment editing and trimming can be numerically specified using options such as --mafft-algorithm, --mafft-op, --mafft-ep, --trimal-algorithm, --trimal-gt, and --allow-innertrimming, with outputs available for comparison. Subjective criteria for species delimitation, such as branch length and bootstrap support, can be uniformly defined using the --collapsedistcutoff and --collapsebscutoff options. The --continue option enables users to reproduce phylogenetic analyses and objectively assess the impact of different variables by modifying options in subsequent runs.
Standardization can also contribute to reproducible phylogenetic analysis. Being able to retrieve intermediate data, such as sequence, alignment, and phylogeny, is crucial to reproducing fungal taxonomic studies (Hibbett et al., 2016). Issues including human errors, the absence of intermediate information, and insufficient method descriptions (i.e. alignment editing, trimming criteria, and species range decision) have plagued fungal taxonomy, reducing the reproducibility of fungal phylogenetic analysis (Petersen et al., 2010; Shen et al., 2020). FunVIP addresses these challenges by consolidating all the step-by-step results into a single data directory. Researchers using FunVIP can easily deposit results as supplementary files, enabling others to reproduce the entire analysis with a single command.
Practical problems arising in fungal taxonomy were also considered in developing FunVIP. Large-scale insertions and fragmented sequences pose challenges in managing fungal sequence alignments. For instance, large-scale insertions are critical criteria for certain fungal species identification (Nagy et al., 2012). In the case of the genus Trichoderma, inconsistent transcription elongation factor regions are used by sequences (e.g. EU338335 contains intron 4 and 5 while DQ835489 includes exon 6), resulting in failing alignments (Dou et al., 2020; Rahimi et al., 2021). To address these alignment issues, FunVIP applies two solutions. Firstly, FunVIP uses the local alignment algorithm of MAFFT as default, instead of the widely used global alignment (Shen et al., 2021), enhancing performance with fragmented sequences or large-scale insertions (Thompson et al., 1999). Secondly, FunVIP preserves the non-terminal columns of the alignment, overcoming a limitation that may inadvertently remove phylogenetically important columns (Supple Data 3.1). Users can efficiently detect and manage such issues by combining these two solutions with the invalid alignment validation process (Supple Data 3.2).
Low-resolution genetic markers from closely related fungal species, such as the ITS region of Penicillium and Aspergillus, can produce polytomies in phylogenetic trees (Houbraken et al., 2021). Traditional tree-inferring tools, such as RAxML, only allow bifurcating trees, where the topology of the clades among identical sequences is not collapsed as intended. To overcome this, a polytomy-resolving algorithm and flat issue reporting system are integrated into FunVIP (Supple Data 8, Fig. S8.1). FunVIP reorganizes the topology of clades including zero-length branches based on annotation evidence. During these processes, FunVIP highlights issues that users should be cautious about. It is expected to facilitate a clearer interpretation of the phylogenetic tree when the genetic marker has low resolution.
For efficient tree inference, FunVIP employs the FastTree algorithm in the fast mode, replacing the commonly used neighbor-joining (NJ) method (Tamura et al., 2004). FastTree is recognized for balancing speed and accuracy, surpassing NJ methods (Price et al., 2010), and yielding results more closely coinciding with maximum likelihood (ML) methods like RAxML compared to the NJ method (Liu et al., 2011). Comparison of identification performance between FastTree (fast mode) and RAxML (accurate mode) with the Sanghuangporus and Aspergillus section Terrei datasets showed a small difference in accuracy, but a large decrease in time consumption (Sanghuangporus-fast: 56s, Sanghuangporus-accurate: 4,420s, Aspergillus section Terrei-fast: 55s, Aspergillus section Terrei-accurate: 4,767s). Therefore, running with FunVIP fast preset can be a good initial identification method, as it has a lower disparity with the ML method compared to the NJ method.
Validation performance of FunVIP from the Fuscoporia dataset
Taxonomic reference data and sequences from public databases are considered as ground truth and used for species identification (Lücking et al., 2020a). However, mislabeled sequences are common in both public sequence databases (Hofstetter et al., 2019) and taxonomic reference data (Cho et al., 2023), leading to misidentifications (Lücking et al., 2020b). Exploring and interpreting all references used for the dataset can be difficult for users. Taxonomic experts can aid in curating these issues, but they are not always available. Therefore, FunVIP is designed to mimic the methods and expertise of taxonomists, to streamline and improve the accuracy of species identification.
Resolving mislabels is one of the most crucial steps in phylogeny-based identification using a public database containing misannotated sequences. One piece of evidence of mislabels is database conflicts: when samples within the database conflict with each other, it indicates that either of them is likely mislabeled. FunVIP detected 113 database conflicts in the Fuscoporia dataset, including taxonomic mislabels and minor conflicts. Most of the database conflicts found in the Fuscoporia dataset resulted from old names, orthographic variants, additional sequencing, and mistakes in uploading sequences to GenBank. Notably, these conflicts are a consequence of the rapid advancement of fungal taxonomy research (Dayarathne et al., 2016). FunVIP validated six name updates in subsequent references, such as F. wahlbergii re-identified as F. australasica, and four orthographic variants, such as F. setifera being misspelled as F. setifer. The other evidence to find mislabels is polyphyly. Multiple clades identified as the same species (polyphyly) usually do not make sense from an evolutionary point of view. Thus, polyphyly should be primarily checked by validating mislabeled samples (Kozlov et al., 2016; McDonald et al., 2012). FunVIP detected 108 instances polyphyly and provides a detailed evidence report to assist users in dataset validation by indicating “where to start” and “what to check”, thus offering a solution when taxonomic expertise is absent. In previous work by Kozlov et al. (2016), the Semi-Automatic Taxonomic Improvement and Validation Algorithm (SATIVA) employed a polyphyly-based approach to curate taxonomically mislabeled samples. SATIVA used the Evolutionary Placement Algorithm (EPA) (Berger et al., 2011) to address polyphyly issues. However, EPA may yield incorrect results if a significant portion of samples are mislabeled, a critical concern in fungal phylogeny [i. e. F. gilva from Cho et al. (2023)]. Additionally, the primary object of SATIVA is to curate databases used in metabarcoding methods. Therefore, some features required by the field of fungal taxonomy, such as full automation (SATIVA starts with alignment), multigene applicability, and identification ability, are not supported. Now, FunVIP meets the demand for a validation program similar to SATIVA in the field of fungal taxonomy.
FunVIP implements a sequence-set organization step to address extreme cases of mislabeled samples included in the database. The sequence-set organization step played a notable role in distinguishing mislabeled samples at the taxonomic group (genus) level. Typically, after taxonomic group designation with BLAST, only ingroup and outgroup samples are included for phylogenetic analysis (Dissanayake et al., 2020). However, FunVIP included ambiguous samples (Fomes, Fomitiporia, Fulvifomes, Inonotus, Inocutis, Phellinopsis, Phellinidium, Phellinus, and Tropicoporus) in the sequence-set when analysing the Fuscoporia dataset. This approach facilitated the detection of non-Fuscoporia samples incorrectly labeled as Fuscoporia samples, such as F. coronadensis (Fig. 3F).
As seen in the Fuscoporia example, the prevalence of taxonomically misidentified sequences in public databases presents a significant challenge, highlighting the need to correct these annotations to improve data quality used by the scientific community. Curating the annotations of these sequences offers a fundamental solution that benefits all researchers utilizing these resources. While GenBank and UNITE have systems in place to address major misidentifications (such as those at the phylum level), minor cases (such as those within the genus level) are either not open for correction in GenBank or manually curated through third-party annotation platforms like UNITE and PlutoF (Abarenkov et al., 2024). FunVIP facilitates the identification and confident evaluation of these minor misidentifications, streamlining the correction process. By combining FunVIP with third-party annotation systems, users can actively contribute their findings, enhancing the accuracy of public sequence databases. This collaborative effort is expected to significantly reduce errors across datasets, ultimately reducing the workload of the community.
Identification performance of FunVIP compared with BLAST
NCBI BLAST is a widely used method for initial fungal identification. However, the identification accuracy of NCBI BLAST in our analysis ranged from 87.2% to 95.1%, which is too low to be confident. BLAST-based fungal identification has several limitations: susceptibility to mislabeled sequences in the database and inconsistencies in similarity cutoffs for delimiting fungal species (Johnson et al., 2008; Somervuo et al., 2016). In addition, the local alignment algorithm of BLAST can reflect only aligned region of the sequences, thus similarity criteria can lead to unexpected result (Kang et al., 2010; Nilsson et al., 2012). We implemented a validation ability in FunVIP that specifies potentially misannotated database samples based on polyphyletic clades. Furthermore, the species delimitation algorithm, which utilizes tree metrics (branch length and branch support), resolves issues related to inconsistent similarity cutoffs.
Inconsistencies in similarity cutoffs for delimiting fungal species, ranging from 0.93 to 1 in similarity scores in the ITS regions, pose a challenge (Vu et al., 2022). BLAST analysis exhibited significantly larger variations in accuracy, precision, recall, and F1 score, depending on the percent identity cutoff settings used (Supple Data 7). In contrast, FunVIP utilizes phylogenetic topology and branch length to capture the range of sequence variation for each species, which is reported as a better approach in cases involving mislabeled samples in the database (Ross et al., 2008). As a result, FunVIP demonstrates lower deviations in identification metrics, ensuring a more consistent outcome irrespective of user-applied options. Additionally, FunVIP can delimitate new species candidates, unlike NCBI BLAST and q2-feature-classifier.
Limitations and prospects
FunVIP can validate potentially mislabeled data but cannot confirm which data is incorrect without additional information, such as geographical origin or morphological characteristics of the studied specimen or isolate. Moreover, decisions are often based on the type specimens, making species without a sequence from the type specimen problematic. Since FunVIP is designed as a pipeline for fungal identification, it may not cover all scenarios, such as manual alignment editing, genetic marker partitioning, and Bayesian analysis.
Although FunVIP was initially developed to address the specific challenges of accurate and efficient fungal identification, its theoretical adaptability extends beyond fungi. However, the algorithms were specifically tailored for fungal data —for instance, by preventing internal column trimming and reorganizing trees for low-resolution barcodes. Since the development of FunVIP was based on expertise in fungal systems, potential challenges specific to other taxa remain unaddressed. Expanding the usability of FunVIP as a universal tool requires rigorous testing by specialists in other taxa to identify and resolve these challenges. To improve user-friendliness, future versions of FunVIP are expected to incorporate additional features such as graphical user interfaces and web servers, enhancing the accessibility and versatility of the pipeline.
Conclusion
Accurate sequence-based identification of fungal species is always in demand. We released FunVIP, a phylogeny-based validation and identification pipeline for fulfilling the demand in fungal identification. This pipeline offers distinctive features: automatic sequence download, detection of misannotated samples in the database, multi-genetic marker analysis, and interpretation of phylogenetic trees. Through three case studies using Fuscoporia, Sanghuangporus, and Aspergillus section Terrei datasets, FunVIP effectively validated mislabeled sequences from databases and demonstrated superior identification performance compared to the widely employed BLAST. Overall, FunVIP serves as a straightforward identification pipeline when a database is available, enhancing confidence in data accuracy and reducing the time required for iterative phylogenetic analysis.
Acknowledgments
This research was supported by the [National Research Foundation of Korea (NRF) funded by the Korean government (MSIT)] under Grant [No. 2023R1A2C1005130]; and [“research on the diversity of indigenous soil microbial species” project of the National Institute of Biological Resources under the Ministry of Environment of the Republic of Korea] under Grant [Nos. NIBR202304107 and NIBR202402106].
We express our appreciation to Ki Hyeong Park (National Institute of Forest Science, KR), Hyunho Moon (Seoul National University, School of Biological Sciences, KR), and Seongjun Kim (Seoul National University, School of Biological Sciences, KR) for assistance in debugging during the FunVIP development. We express our appreciation to Dong Wook Kim (Seoul National University, Interdisciplinary Program in Bioinformatics, KR) for assistant in managing UFCG dataset for algorithm validation. We also express our appreciation to Sari Timonen (University of Helsinki, FI) for revising the manuscript.
FunVIP is distributed under GNU GPL3 license. The source code of FunVIP is available at https://github.com/Changwanseo/FunVIP, and archived at Zenodo repository, https://doi.org/10.5281/zenodo.14662698. Pypi repository of FunVIP is available at https://pypi.org/project/FunVIP/. All analysis scripts and files to regenerate analysis in the article is available at Zenodo repository, https://doi.org/10.5281/zenodo.14650006.
Conflict of Interest
The authors declare that they have no competing interests.
Supplementary Information
The online version contains supplementary material available at https://doi.org/10.71150/jm.2411017.
Supple Data 7.
Mean and standard deviation values for accuracy, precision, recall, and F1 score of FunVIP, BLAST, and q2-feature-classifier using the Sanghuangporus and Aspergillus section Terrei datasets
jm-2411017-Supplementary-Data-7.docx
Fig. 1.FunVIP workflow and usage. (A) Schematic representation of the phylogenetic tree-based identification using FunVIP. The red arrows indicate validation checkpoints in each process, and the blue arrow indicates the FunVIP pipeline. Small letters denote each genetic marker. (B) Database and query input format example with descriptions for FunVIP. Columns marked with red asterisks are mandatory to fill for the database file, and optional for the query file. (C) Example command and its description to execute FunVIP.
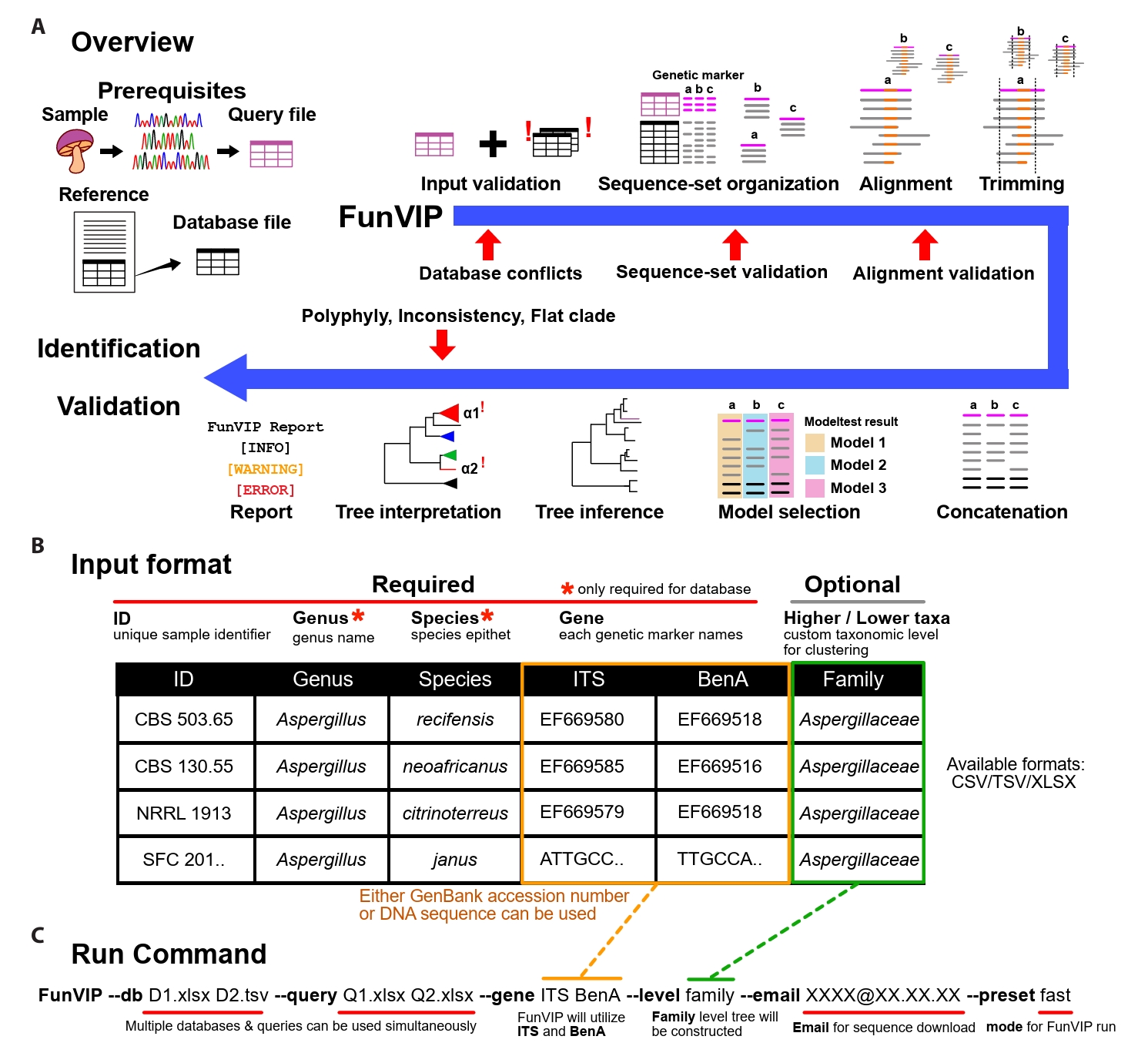
Fig. 2.Tree interpretation algorithms performed in FunVIP. Infographics describing algorithms performed during the tree interpretation step. Taxa α, β, and γ indicate database samples, O indicates database samples selected as the outgroup, and x, y, and z indicate query samples. Red, blue, and green colors indicate taxa α, β, and γ respectively. (A) Rooting of outgroup. Crimson squares indicate the outgroup. (B) Polytomy-resolving and monophyly detection. The crimson square indicates polytomy of taxon γ, and dotted squares indicate species’ borders defined by monophyly detection. The bidirectional arrow indicates recursive calls between the two functions. (C) Metric (tree distance, bootstrap)-based clustering. Dotted squares indicate species’ borders adjusted by metric-based clustering. MRCA stands for the Most Recent Common Ancestor. (D) Collapsing. (E) Synchronizing across each genetic marker tree. The crimson square indicates different criteria applied for collapsing by each genetic marker tree. Purple circles indicate sample z assigned to different species by each genetic marker tree. (F) Visualizing and reporting.
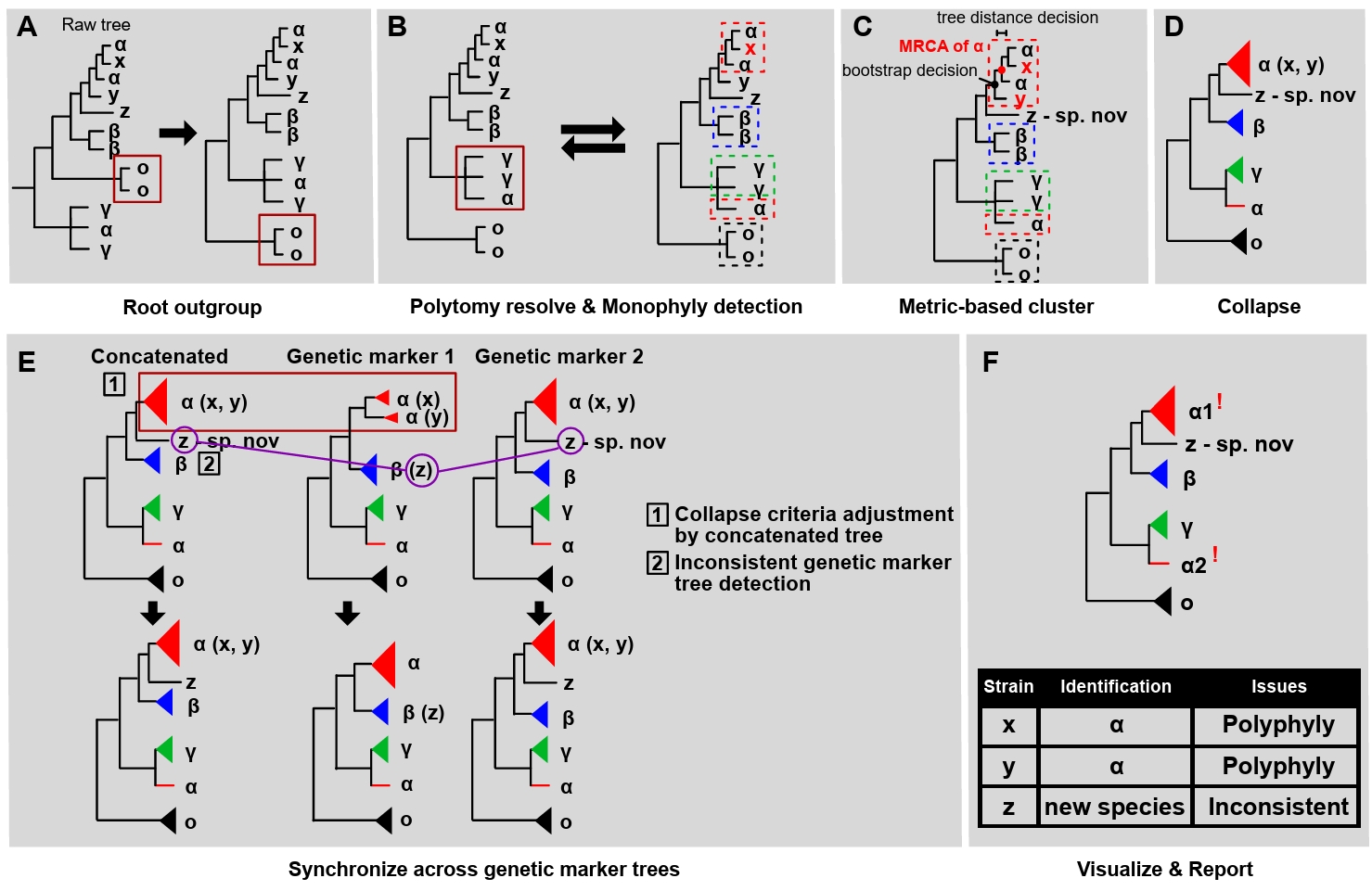
Fig. 3.Validation ability of FunVIP on re-analyzing the Fuscoporia dataset. Mint-colored squares indicate mislabeled taxonomy and orange-colored squares indicate mislabeled genetic markers. (A) The initial FunVIP run failed due to database conflicts. (B) Phylogenetic tree generated with polyphyly issues detected by FunVIP using the curated Fuscoporia database. (C) Mislabeled taxonomy detected from database conflicts. Red squares indicate conflicting species epithets among the samples in the database. (D) Mislabeled sample found by polyphyly at the species level. The red square indicates true F. gilva, and the blue squares indicate mislabeled F. gilva. (E) Mislabeled sample found by polyphyly at the taxonomic group (genus) level. (F) Samples with mislabeled genetic markers (ITS and LSU switched) were found by polyphyly. The black color in the alignment indicates unaligned regions to consensus, and the grey color in the alignment indicates aligned regions to consensus. Red squares indicate LSU sequences mislabeled as ITS.
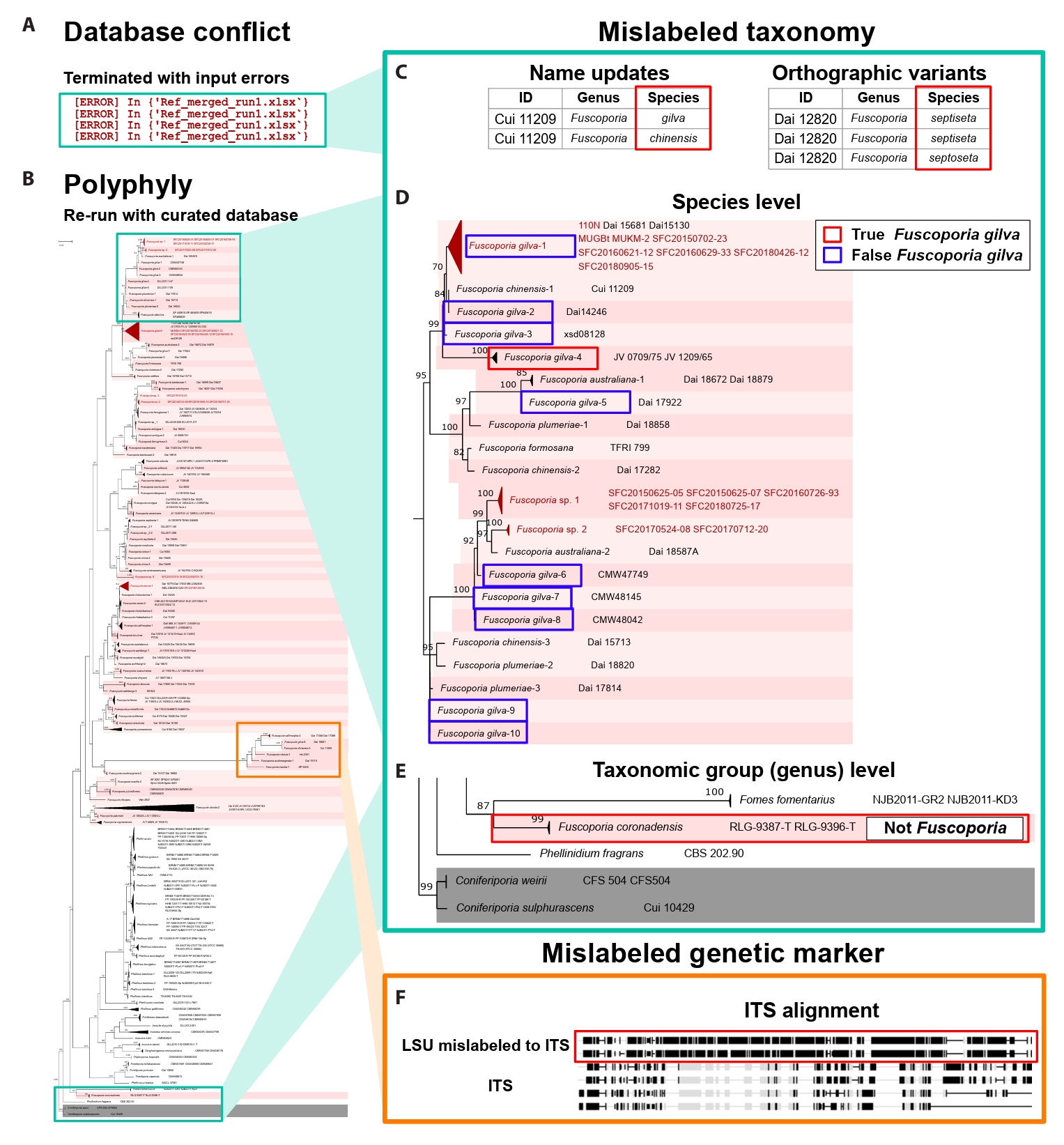
Fig. 4.Accuracy, precision, recall, and F1 score for FunVIP, BLAST, and q2-feature-classifier using the Sanghuangporus and Aspergillus section Terrei datasets. (A) Multi-classifier metrics of the Sanghuangporus dataset using the ITS region. (B) Multi-classifier metrics of the Aspergillus section Terrei dataset with the multiple genetic markers, BenA and CaM. The analyses included FunVIP-fast, FunVIP-accurate, BLAST, and q2-feature-classifier. Individual data points of the analyses are represented by dots. The X-axes indicate options that were applied to each of the analyses (FunVIP – collapsedistcutoff, BLAST – percent identity cutoff, q2-feature-classifier – confidence threshold). The Y-axes indicate each of the metrics in the title of the graphs.
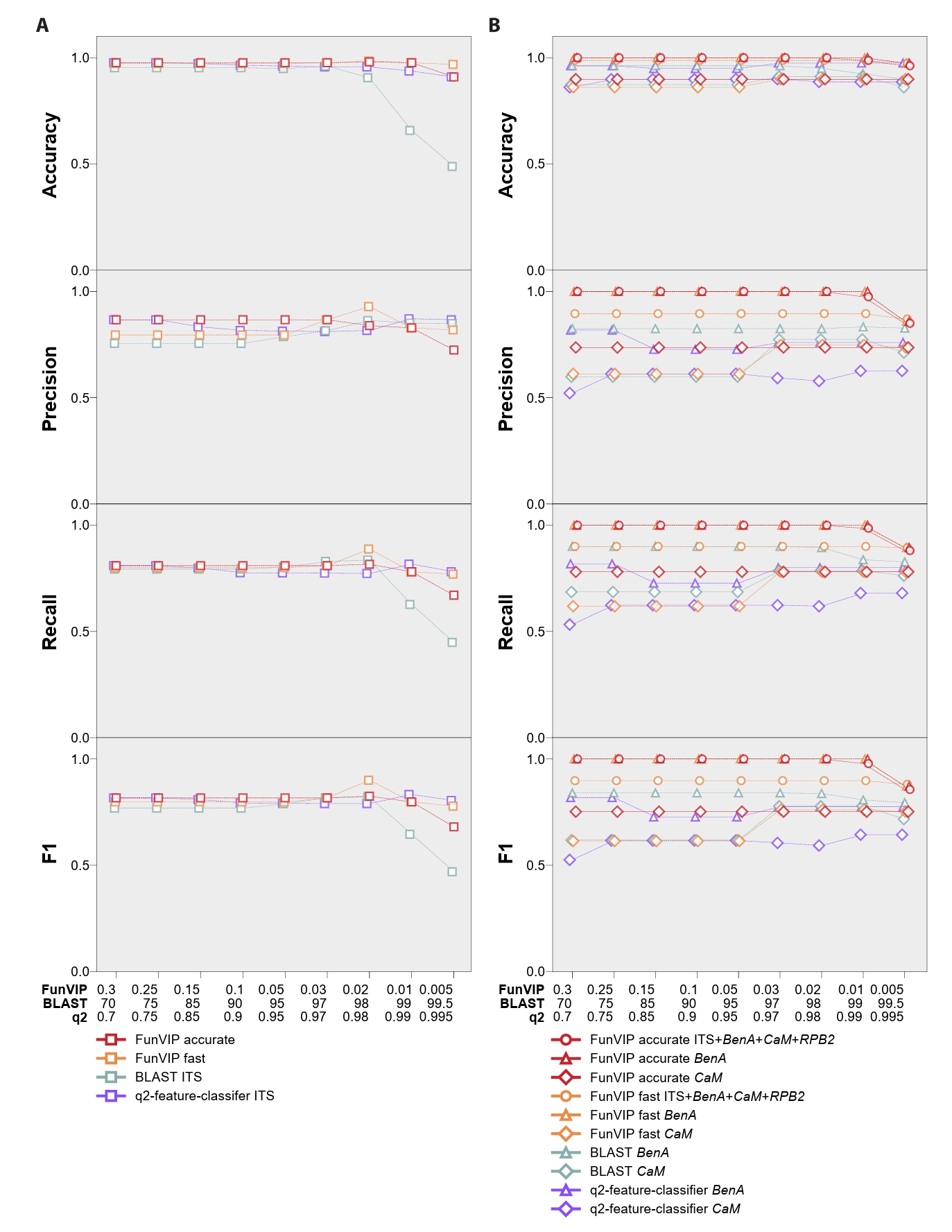
Table 1.Comparison of potential softwares for fungal identification
|
Software |
Key features |
Limitations in fungal identification |
Intermediate result availability |
Final identification result availability |
Multiple genetic marker applicability |
Reference |
|
Phylogeny.fr |
Customizable phylogeny-based identification |
Requires user interpretation of tree outcomes |
Yes |
No |
Yes |
Dereeper et al. (2008)
|
|
One-click Fungal Phylogenetic Tool (OFPT) |
Streamlined fungal phylogenetic analysis |
Requires user interpretation of tree outcomes |
Yes |
No |
Yes |
Zeng et al. (2023)
|
|
SUPERSMART |
Automated phylogeny-based analysis for established species |
For species delimitation not identification |
Yes |
No |
Yes |
Antonelli et al. (2017)
|
|
OneTwoTree |
Automated phylogeny-based analysis for established species |
For species delimitation not identification |
Yes |
No |
Yes |
Drori et al. (2018)
|
|
SATIVA |
Semi-automatic tool for validating phylogenetic trees |
Prior phylogenetic analysis required |
Yes |
- |
- |
Kozlov et al. (2016)
|
|
q2-feature-classifier |
QIIME 2 plugin for taxonomy classification using k-mer search |
Limited to single marker sequences |
No |
Yes |
No |
Bokulich et al. (2018)
|
|
Does not provide phylogenetic analysis or species identification |
|
BLAST |
Local alignment-based similarity search against database |
Limited to single marker sequences |
No |
No |
Yes |
Camacho et al. (2009)
|
|
Does not provide phylogenetic analysis or species identification |
|
FunVIP |
Fully automated phylogeny-based identification pipeline for fungi |
Currently only tested with fungal datasets |
Yes |
Yes |
Yes |
This study |
Table 2.Number of polyphyly issues and inconsistent issues reported in second FunVIP run with Fuscoporia dataset
|
Issue |
Genetic marker |
Criteria |
Number of issues |
|
Polyphyly |
concatenated |
samples |
108 |
|
clades |
50 |
|
species |
20 |
|
ITS |
samples |
109 |
|
clades |
48 |
|
species |
20 |
|
LSU |
samples |
59 |
|
clades |
33 |
|
species |
14 |
|
RPB2
|
samples |
36 |
|
clades |
14 |
|
species |
7 |
|
TEF1
|
samples |
17 |
|
clades |
9 |
|
species |
6 |
|
Inconsistent |
|
|
89 |
References
- Abarenkov K, Nilsson RH, Larsson KH, Taylor AF, May TW, et al. 2024. The UNITE database for molecular identification and taxonomic communication of fungi and other eukaryotes: sequences, taxa and classifications reconsidered. Nucleic Acids Res. 52(D1): D791–D797. ArticlePubMedPMCPDF
- Alexopoulos CJ, Mims CW, Blackwell M. 1996. Introductory Mycology. 4th edn. Wiley
- Anslan S, Nilsson RH, Wurzbacher C, Baldrian P, Leho Tedersoo, et al. 2018. Great differences in performance and outcome of high-throughput sequencing data analysis platforms for fungal metabarcoding. MycoKeys. (39): 29–40.ArticlePubMedLink
- Antonelli A, Hettling H, Condamine FL, Vos K, Nilsson RH, et al. 2017. Toward a self-updating platform for estimating rates of speciation and migration, ages, and relationships of taxa. Syst Biol. 66(2): 152–166. ArticlePubMedPMC
- Atkins SD, Clark IM. 2004. Fungal molecular diagnostics: a mini review. J Appl Genet. 45(1): 3–15. PubMed
- Barros Correia AC, Barbosa RN, Frisvad JC, Houbraken J, Souza-Motta CM. 2020. The polyphasic re-identification of a Brazilian Aspergillus section Terrei collection led to the discovery of two new species. Mycol Prog. 19: 885–903. ArticlePDF
- Berger SA, Krompass D, Stamatakis A. 2011. Performance, accuracy, and web server for evolutionary placement of short sequence reads under maximum likelihood. Syst Biol. 60(3): 291–302. ArticlePubMedPMC
- Bhunjun CS, Niskanen T, Suwannarach N, Wannathes N, Chen YJ, et al. 2022. The numbers of fungi: are the most speciose genera truly diverse? Fungal Divers. 114(1): 387–462. ArticlePDF
- Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, et al. 2018. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome. 6: 1–17. ArticlePubMedPMCPDF
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, et al. 2009. BLAST+: architecture and applications. BMC Bioinformatics. 10: 1–9. ArticlePubMedPMCPDF
- Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 25(15): 1972–1973. ArticlePubMedPMCPDF
- Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 17(4): 540–552. ArticlePubMed
- Chang JM, Di Tommaso P, Notredame C. 2014. TCS: a new multiple sequence alignment reliability measure to estimate alignment accuracy and improve phylogenetic tree reconstruction. Mol Biol Evol. 31(6): 1625–1637. ArticlePubMed
- Chase MW, Fay MF. 2009. Barcoding of plants and fungi. Science. 325(5941): 682–683. ArticlePubMed
- Chen Q, Dai YC. 2019. Two new species of Fuscoporia (Hymenochaetales, Basidiomycota) from southern China based on morphological characters and molecular evidence. MycoKeys. 61: 75–89. ArticlePubMedPMCLink
- Cho Y, Kim D, Lee Y, Jeong J, Hussain S, et al. 2023. Validation of Fuscoporia (Hymenochaetales, Basidiomycota) ITS sequences and five new species based on multi-marker phylogenetic and morphological analyses. IMA Fungus. 14: 12.ArticlePubMedPMCPDF
- Darriba D, Posada D, Kozlov AM, Stamatakis A, Morel B, et al. 2020. ModelTest-NG: a new and scalable tool for the selection of DNA and protein evolutionary models. Mol Biol Evol. 37(1): 291–294. ArticlePubMedPMCPDF
- Dayarathne MC, Boonmee S, Braun U, Crous PW, Daranagama DA, et al. 2016. Taxonomic utility of old names in current fungal classification and nomenclature: Conflicts, confusion & clarifications. Mycosphere. 7(11): 1622–1648. Article
- Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, et al. 2008. Phylogeny. fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36(Web Server issue): W465–W469. ArticlePubMedPMC
- Dissanayake AJ, Bhunjun CS, Maharachchikumbura SSN, Liu JK. 2020. Applied aspects of methods to infer phylogenetic relationships amongst fungi. Mycosphere. 11(1): 2652–2676. Article
- Dou K, Lu Z, Wu Q, Ni M, Yu C, et al. 2020. MIST: A multilocus identification system for Trichoderma. Appl Environ Microbiol. 86(18): e01532–20. ArticlePubMedPMCLink
- Drori M, Rice A, Einhorn M, Chay O, Glick L, et al. 2018. OneTwoTree: An online tool for phylogeny reconstruction. Mol Ecol Resour. 18(6): 1492–1499. ArticlePubMedLink
- Du P, Chen Q, Vlasak J. 2020. Fuscoporia ambigua sp. nov., a new species from America and China. Phytotaxa. 456(2): 175–185. Article
- Frisvad JC, Nielsen KF, Samson RA. 2006. Recommendations concerning the chronic problem of misidentification of mycotoxigenic fungi associated with foods and feeds. Adv Exp Med Biol. 571: 33–46. ArticlePubMed
- Hawksworth DL. 2004. ‘Misidentifications’ in fungal DNA sequence databanks. New Phytol. 161(1): 13–15. ArticleLink
- Hawksworth DL, Kirk PM, Sutton BC, Pegler DN. 1996. Ainsworth’s and Bisby’s Dictionary of the Fungi, 8th ed
- Hawksworth DL, Lücking R. 2017. Fungal diversity revisited: 2.2 to 3.8 million species. Microbiol Spectr. 5(4): 10.1128.ArticlePubMedPMCLink
- Hebert PD, Cywinska A, Ball SL, DeWaard JR. 2003. Biological identifications through DNA barcodes. Proc Biol Sci. 270(1512): 313–321. ArticlePubMedPMC
- Hibbett DS. 1992. Ribosomal RNA and fungal systematics. Trans Mycol Soc Japan. 33(4): 533–556.Link
- Hibbett D, Abarenkov K, Kõljalg U, Öpik M, Chai B, et al. 2016. Sequence-based classification and identification of Fungi. Mycologia. 108(6): 1049–1068. PubMed
- Hibbett DS, Taylor JW. 2013. Fungal systematics: is a new age of enlightenment at hand? Nat Rev Microbiol. 11(2): 129–133. ArticlePubMedPDF
- Hillis DM. 1987. Molecular versus morphological approaches to systematics. Annu Rev Ecol Syst. 18: 23–42. Article
- Hofstetter V, Buyck B, Eyssartier G, Schnee S, Gindro K. 2019. The unbearable lightness of sequenced-based identification. Fungal Divers. 96(1): 243–284. ArticlePDF
- Houbraken J, Visagie CM, Frisvad JC. 2021. Recommendations to prevent taxonomic misidentification of genome-sequenced fungal strains. Microbiol Resour Announc. 10(48): e01074–20. ArticlePubMedPMCLink
- Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S. 2008. NCBI BLAST: a better web interface. Nucleic Acids Res. 36(Web Server issue): W5–W9. ArticlePubMedPMC
- Kalyaanamoorthy S, Minh BQ, Wong TK, Von Haeseler A, Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14(6): 587–589. ArticlePubMedPMCPDF
- Kandawatte TC, Manawasinghe IS, Hurdeal VG, Bhunjun CS, Appadoo MA, et al. 2021. What are fungal species and how to delineate them? Fungal Divers. 109(1): 1–25. ArticlePDF
- Kang S, Mansfield MA, Park B, Geiser DM, Ivors KL, et al. 2010. The promise and pitfalls of sequence-based identification of plant-pathogenic fungi and oomycetes. Phytopathology. 100(8): 732–737. ArticlePubMed
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4): 772–780. ArticlePubMedPMC
- Köhler JR, Hube B, Puccia R, Casadevall A, Perfect JR. 2017. Fungi that infect humans. Microbiol Spectr. 5(3): 10.ArticlePubMedPMC
- Kõljalg U, Larsson KH, Abarenkov K, Nilsson RH, Alexander IJ, et al. 2005. UNITE: a database providing web‐based methods for the molecular identification of ectomycorrhizal fungi. New Phytol. 166(3): 1063–1068. ArticlePubMedLink
- Kozlov AM, Zhang J, Yilmaz P, Glöckner FO, Stamatakis A. 2016. Phylogeny-aware identification and correction of taxonomically mislabeled sequences. Nucleic Acids Res. 44(11): 5022–5033. ArticlePubMedPMC
- Liu K, Linder CR, Warnow T. 2011. RAxML and FastTree: comparing two methods for large-scale maximum likelihood phylogeny estimation. PLoS One. 6(11): e27731.ArticlePubMedPMC
- Lücking R, Aime MC, Robbertse B, Miller AN, Aoki T, et al. 2021. Fungal taxonomy and sequence-based nomenclature. Nat Microbiol. 6(5): 540–548. ArticlePubMedPMCPDF
- Lücking R, Aime MC, Robbertse B, Miller AN, Ariyawansa HA, et al. 2020a. Unambiguous identification of fungi: where do we stand and how accurate and precise is fungal DNA barcoding? IMA Fungus. 11(1): 14.ArticlePubMedPMC
- Lücking R, Hawksworth DL. 2018. Formal description of sequence-based voucherless Fungi: promises and pitfalls, and how to resolve them. IMA Fungus. 9(1): 143–165. ArticlePubMedPMCPDF
- Lücking R, Truong BV, Huong DTT, Le NH, Nguyen QD, et al. 2020b. Caveats of fungal barcoding: a case study in Trametes s. lat. (Basidiomycota: Polyporales) in Vietnam reveals multiple issues with mislabelled reference sequences and calls for third-party annotations. Willdenowia. 50(3): 383–403. Article
- McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, et al. 2012. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6(3): 610–618. ArticlePubMedPMCPDF
- Meiklejohn KA, Damaso N, Robertson JM. 2019. Assessment of BOLD and GenBank-Their accuracy and reliability for the identification of biological materials. PLoS One. 14(6): e0217084. ArticlePubMedPMC
- Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, et al. 2020. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. 37(5): 1530–1534. ArticlePubMedPMCPDF
- Nagy LG, Kocsubé S, Csanádi Z, Kovács GM, Petkovits T, et al. 2012. Re-mind the gap! Insertion-deletion data reveal neglected phylogenetic potential of the nuclear ribosomal internal transcribed spacer (ITS) of fungi. PLoS One. 7(11): e49794. ArticlePubMedPMC
- Nguyen NH, Song Z, Bates ST, Branco S, Tedersoo L, et al. 2016. FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 20: 241–248.Article
- Nilsson RH, Anslan S, Bahram M, Wurzbacher C, Baldrian P, et al. 2019a. Mycobiome diversity: high-throughput sequencing and identification of fungi. Nat Rev Microbiol. 17(2): 95–109. ArticlePubMedPDF
- Nilsson RH, Larsson KH, Taylor AFS, Bengtsson-Palme J, Jeppesen TS, et al. 2019b. The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 47(D1): D259–D264. ArticlePubMedPMC
- Nilsson RH, Ryberg M, Kristiansson E, Abarenkov K, Larsson KH, et al. 2006. Taxonomic reliability of DNA sequences in public sequence databases: a fungal perspective. PLoS One. 1(1): e59. ArticlePubMedPMC
- Nilsson RH, Tedersoo L, Abarenkov K, Ryberg M, Kristiansson E, et al. 2012. Five simple guidelines for establishing basic authenticity and reliability of newly generated fungal ITS sequences. MycoKeys. 4: 37–63. Link
- Notredame C, Higgins DG, Heringa J. 2000. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 302(1): 205–217. ArticlePubMed
- Pearson WR, Lipman DJ. 1988. Improved tools for biological sequence comparison. Proc Natl Acad Sci USA. 85(8): 2444–2448. ArticlePubMedPMC
- Petersen G, Knudsen H, Seberg O. 2010. Alignment, clade robustness and fungal phylogenetics—Crepidotaceae and sister families revisited. Cladistics. 26(1): 62–71. ArticlePubMed
- Price MN, Dehal PS, Arkin AP. 2010. FastTree 2-approximately maximum-likelihood trees for large alignments. PLoS One. 5(3): e9490. ArticlePubMedPMC
- Rahimi MJ, Cai F, Grujic M, Chenthamara K, Druzhinina IS. 2021. Molecular identification of Trichoderma reesei. In Mach-Aigner AR, Martzy R (eds.), Trichoderma reesei: Methods and Protocols, pp. 157-175, SpringerLink
- Raja HA, Miller AN, Pearce CJ, Oberlies NH. 2017. Fungal identification using molecular tools: a primer for the natural products research community. J Nat Prod. 80(3): 756–770. ArticlePubMedPMCLink
- Ross HA, Murugan S, Sibon Li WL. 2008. Testing the reliability of genetic methods of species identification via simulation. Syst Biol. 57(2): 216–230. ArticlePubMed
- Sayers EW, Cavanaugh M, Clark K, Pruitt KD, Sherry ST, et al. 2023. GenBank 2023 update. Nucleic Acids Res. 51(D1): D141–D144. ArticlePubMedPMCPDF
- Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, et al. 2012. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci USA. 109(16): 6241–6246. PubMedPMC
- Seo CW, Kim SH, Lim YW, Park MS. 2022. Re-identification on Korean Penicillium sequences in GenBank collected by software GenMine. Mycobiology. 50(4): 231–237. ArticlePubMedPMC
- Shen XX, Li Y, Hittinger CT, Chen XX, Rokas A. 2020. An investigation of irreproducibility in maximum likelihood phylogenetic inference. Nat Commun. 11: 6096.ArticlePubMedPMCPDF
- Shen S, Liu SL, Jiang JH, Zhou LW. 2021. Addressing widespread misidentifications of traditional medicinal mushrooms in Sanghuangporus (Basidiomycota) through ITS barcoding and designation of reference sequences. IMA Fungus. 12: 10.ArticlePubMedPMCPDF
- Sokoł S, Kaldorf M, Bothe H. 1999. Molecular characterization and taxonomic affinities of species of the white rot fungus Ganoderma. Z Naturforsch C J Biosci. 54(5-6): 314–318. ArticlePubMed
- Somervuo P, Koskela S, Pennanen J, Henrik Nilsson R, Ovaskainen O. 2016. Unbiased probabilistic taxonomic classification for DNA barcoding. Bioinformatics. 32(19): 2920–2927. ArticlePubMedPDF
- Spivak ES, Hanson KE. 2018. Candida auris: an emerging fungal pathogen. J Clin Microbiol. 56(2): e01588–17. ArticlePubMedPMCLink
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9): 1312–1313. ArticlePubMedPMCPDF
- Steinegger M, Söding J. 2017. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat Biotechnol. 35(11): 1026–1028. ArticlePubMedPDF
- Tamura K, Nei M, Kumar S. 2004. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci USA. 101(30): 11030–11035. ArticlePubMedPMC
- Taylor JW. 2011. One fungus = one name: DNA and fungal nomenclature twenty years after PCR. IMA Fungus. 2: 113–120. ArticlePubMedPMCPDF
- Tedersoo L, Bahram M, Põlme S, Kõljalg U, Yorou NS, et al. 2014. Global diversity and geography of soil fungi. Science. 346(6213): 1256688.PubMed
- Tedersoo L, Bahram M, Zinger L, Nilsson RH, Kennedy PG, et al. 2022. Best practices in metabarcoding of fungi: from experimental design to results. Mol Ecol. 31: 2769–2795.ArticlePubMedLink
- Thompson JD, Plewniak F, Poch O. 1999. A comprehensive comparison of multiple sequence alignment programs. Nucleic Acids Res. 27(13): 2682–2690. ArticlePubMedPMC
- Tomé LMR, Badotti F, Assis GBN, Fonseca PLC, da Silva GA, et al. 2019. Proteomic fingerprinting for the fast and accurate identification of species in the Polyporoid and Hymenochaetoid fungi clades. J Proteomics. 203: 103390.ArticlePubMed
- Vu D, Nilsson RH, Verkley GJ. 2022. Dnabarcoder: An open‐source software package for analysing and predicting DNA sequence similarity cutoffs for fungal sequence identification. Mol Ecol Resour. 22(7): 2793–2809. ArticlePubMedPMC
- Wei Y, Li L, Liu Y, Xiang S, Zhang H, et al. 2022. Identification techniques and detection methods of edible fungi species. Food Chem. 374: 131803.ArticlePubMed
- White TJ, Bruns T, Lee S, Taylor J. 1990. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In Innis MA, Gelfand DH, Sninsky JJ, White TJ. (eds.), PCR Protocols: A Guide to Methods and Applications, vol. 18, pp. 315–322. Academic Press, Inc.
- Wu F, Zhou LW, Vlasák J, Dai YC. 2022. Global diversity and systematics of Hymenochaetaceae with poroid hymenophore. Fungal Divers. 113(1): 1–192. ArticlePDF
- Zeng XY, Tan TJ, Tian FH, Wang Y, Wen TC. 2023. OFPT: A one-stop software for fungal phylogeny. Mycosphere. 14(1): 1730–1741. Article
Citations
Citations to this article as recorded by

- Hidden diversity of crust-like Sebacinaceae (Sebacinales, Agaricomycetes) in Asia
Hannah Suh, Chang Wan Seo, Ki Hyeong Park, Shinnam Yoo, Dohye Kim, Yoonhee Cho, Young Woon Lim
IMA Fungus.2026;[Epub] CrossRef - The contribution of environmental DNA to exploring hypogeous fungal diversity and vulnerability
Chang Wan Seo, Shinnam Yoo, Hannah Suh, Dohye Kim, Hyun Lee, Young Woon Lim
BMC Microbiology.2026;[Epub] CrossRef - Development of molecular assays to detect the G143A point mutation responsible for group 11 fungicide insensitivity in Colletotrichum lentis
Zakir Hossain, Michelle Hubbard
Journal of Plant Pathology.2026; 108(2): 1255. CrossRef -
Cryptoporus densiflorus
, sp. nov. (Polyporaceae), from East Asia and a reassessment of species distributions in
Cryptoporus
Jiyun Choi, Abel Severin Lupala, Ji Seon Kim, Yu-Cheng Dai, Young Woon Lim
Mycologia.2026; : 1. CrossRef - Exploring Macrofungal Biodiversity and Distribution on Kyodong Island, Republic of Korea
Hannah Suh, Abel Severin Lupala, Hae Jin Cho, Sumin Jo, Jiyun Choi, Young Woon Lim
Mycobiology.2025; 53(4): 466. CrossRef -
Expanding the Inventory of Seven Unrecorded Marine
Penicillium
with Morphological Descriptions and Phenotypic Variability
Wonjun Lee, Ji Seon Kim, Sumin Jo, Young Woon Lim
Mycobiology.2025; 53(5): 648. CrossRef -
Exploring Fungal Diversity in Marine Plastic (PET) Wastes and Seafoam in Udo Island, South Korea, with Reports of Two New Species (
Leptospora conidiifera
and
Neodevriesia oceanoplastica
)
Wonjun Lee, Sumin Jo, Soo Hyun Maeng, Ji Seon Kim, Myung Kyum Kim, Young Woon Lim
Mycobiology.2025; 53(6): 770. CrossRef - Potential of Trichoderma asperellum against root-rot caused by Fusarium equiseti in tomato plants
Louis Antoniel Joseph, Manoucheca Jean, Frantzdy Luc, Kerley-Vivaldi Jean, Bento Gil Uane, Marisa Aida Diogo Matsinhe, Meque Samuel Tivane, Inocêncio Oliveira Mulaveia
Research, Society and Development.2025; 14(12): e62141250223. CrossRef


 ePub Link
ePub Link Cite this Article
Cite this Article