ABSTRACT
- Dual specificity phosphatases (DUSPs) are a subfamily of protein tyrosine phosphatases that regulate diverse cellular processes through dephosphorylation of phosphorylated substrates. DUSPs are commonly found in eukaryotes, bacteria, archaea, and viruses. However, structural and biochemical characterization of bacterial DUSP remains limited, as only one bacterial DUSP has been identified thus far. In this study, we investigated a novel putative bacterial DUSP from Candidatus Chlorohelix allophototropha, referred to as CCaDUSP. The crystal structure of CCaDUSP showed the presence of a well-conserved catalytic motif with a characteristic phosphate-binding loop. Biochemical analyses further confirmed that CCaDUSP exhibits phosphatase activity and contains dual general acid/base residues, both of which contribute to its enzymatic activity. These findings not only represent the first characterization of a novel bacterial DUSP with dual general acid/base residues but also provide a foundation for understanding the diversity of DUSP proteins in bacteria.
-
Keywords: dual specificity phosphatase, protein tyrosine phosphatase, DUSP, PTP, Candidatus Chlorohelix allophototropha, CCaDUSP, crystal structure
Introduction
Protein phosphorylation is a key post-translational modification involved in the regulation of various cellular processes. Cellular phosphorylation levels are tightly controlled by the coordinated action of kinases and protein phosphatases (Tonks, 2006). The protein tyrosine phosphatase (PTP) family consists of diverse enzymes that catalyze the removal of phosphate groups from phosphorylated substrates (Alonso et al., 2004). Dual specificity phosphatases (DUSPs) constitute a subfamily of PTPs that are capable of dephosphorylating both phosphoserine/phosphothreonine and phosphotyrosine residues (Patterson et al., 2009). DUSP proteins possess a conserved catalytic domain containing the signature phosphate-binding loop (P-loop) motif, H−C−x−x−G−x−x−R−S/T (Jeong et al., 2014). DUSPs constitute the sole PTP subfamily commonly found in eukaryotes, bacteria, archaea, and viruses (Ku, 2025). To date, only one bacterial DUSP, TpbA from Pseudomonas aeruginosa, has been structurally and biochemically characterized (Koveal et al., 2013; Xu et al., 2015). Recently, we reported the presence of an additional putative bacterial DUSP protein in Candidatus Chlorohelix allophototropha (Ku, 2025), an anoxygenic phototroph in the Chloroflexota phylum (Tsuji et al., 2024). Sequence alignment of this protein, referred to as CCaDUSP in this study, suggests that it shares similarities with several representative DUSP family members, including P. aeruginosa TpbA (a bacterial DUSP), Thermococcus kodakaraensis Tk-PTP (an archaeal DUSP), and human DUSP23a (Fig. 1A). In the present study, we investigated the crystal structure and enzymatic characteristics of CCaDUSP. We demonstrated that it harbors a well-conserved catalytic motif and dual general acid/base residues, providing a basis for understanding the diversity of DUSP family proteins in bacteria.
Materials and Methods
Preparation, crystallization, and structural determination
The coding region corresponding to residues 1–160 of CCaDUSP (RefSeq: WP_341469495.1) was subcloned into a modified pET28a vector (Novagen) fused in frame with an His10−maltose-binding protein tag at the N-terminal region. The CCaDUSP recombinant protein was expressed in Escherichia coli BL21(DE3) RIL cells (Novagen) cultured in Luria–Bertani medium at 25°C for 12 h following induction with 0.1 mM isopropyl β-D-thiogalactopyranoside. The protein was purified using Ni-NTA affinity chromatography (QIAGEN), followed by size-exclusion chromatography on a HiLoad 26/600 Superdex 75 prep grade column (Cytiva). The His10−maltose-binding protein tag was removed during purification after cleavage with tobacco etch virus protease. Protein samples were equilibrated with running buffer containing 50 mM Tris–HCl (pH 7.5), 200 mM NaCl, and 2 mM dithiothreitol. Crystallization trials were set up using the sitting-drop vapor diffusion method at 18°C by mixing 1 μl of protein solution (20 mg/ml) with 1 μl of precipitant solution, which is composed of 0.1 M Tris–HCl (pH 8.5), 1.5 M ammonium sulfate, and 12% glycerol. Diffraction data were collected at Beamline 5C of the Pohang Accelerator Laboratory (Korea) and processed using HKL2000 (Otwinowski and Minor, 1997). Molecular replacement was carried out using Phaser (McCoy et al., 2007) with an AlphaFold3-predicted model and crystal structures of TpbA (Koveal et al., 2013; Xu et al., 2015), Tk-PTP (Yun et al., 2018), DUSP23a (Agarwal et al., 2008; Kuznetsov et al., 2012), and DUSP28 (Ku et al., 2017) as search models (Table 1). The molecular replacement solution obtained using the AlphaFold3-predicted model was selected for further analysis based on the log-likelihood gain (LLG) and translation-function Z-score (TFZ) values (Table 1). Model building and refinement were performed using Coot (Emsley and Cowtan, 2004) and PHENIX (Adams et al., 2010), respectively. The crystallographic statistics are presented in Table 2.
Dephosphorylation activity assays
The wild-type construct served as a template for preparing CCaDUSP mutants containing D67N, C97S, or E135Q substitutions. These proteins were prepared following a procedure similar to that used for the wild-type protein. The enzymatic activity of CCaDUSP was measured using para-nitrophenylphosphate (pNPP) as a substrate. To determine the optimal pH for the enzymatic reaction, the initial reaction buffers containing 50 mM NaCl, 2 mM dithiothreitol, and 100 mM Bis-Tris adjusted to pH values ranging from 5.5 to 8.0 were prepared. Subsequent enzymatic analyses were performed at pH 5.5, which was optimal for this protein. For the kinetic analysis, the reaction mixture containing 20 μM recombinant wild-type or mutant CCaDUSP was incubated with varying concentrations of pNPP (0–90 mM). The reaction was carried out for 5 min and then immediately terminated by adding 80 μl of 1 N NaOH. The mixture was allowed to stand for 30 min to stabilize the chromophores. The substrate hydrolysis rate was then determined by measuring the absorbance at 405 nm using a SpectraMax Plus 384 spectrophotometer (Molecular Devices, USA) with its chamber temperature set at 30°C. To express the kinetic rates in quantitative units, the raw velocity (Abs/min) was converted to nmol/min based on the Beer–Lambert law, using the molar extinction coefficient of p-nitrophenol (ε = 18,000 M-1 cm-1) and the optical pathlength (b = 0.45 cm). All enzymatic assays were performed in triplicate.
Results and Discussion
Crystal structure of CCaDUSP
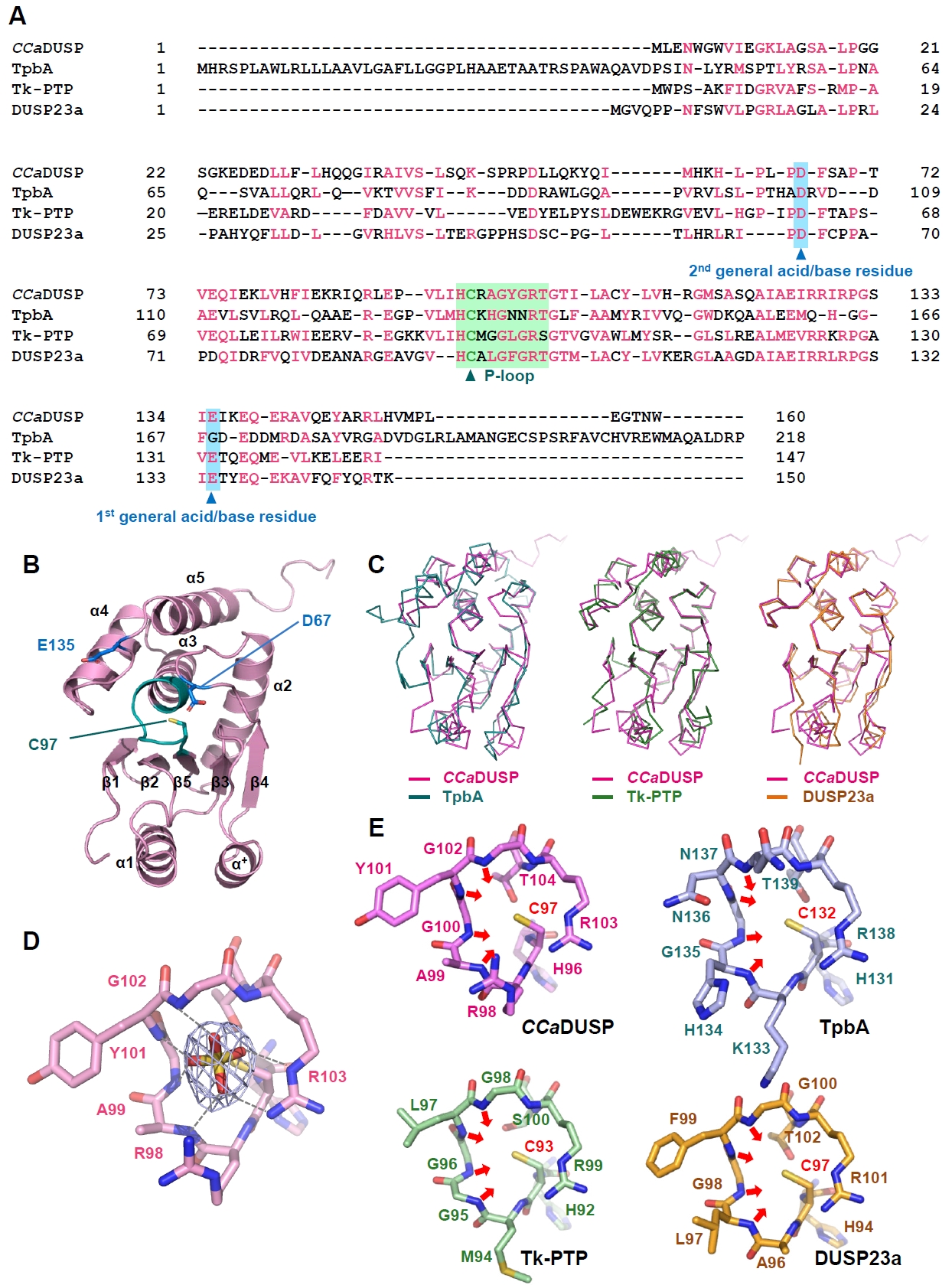
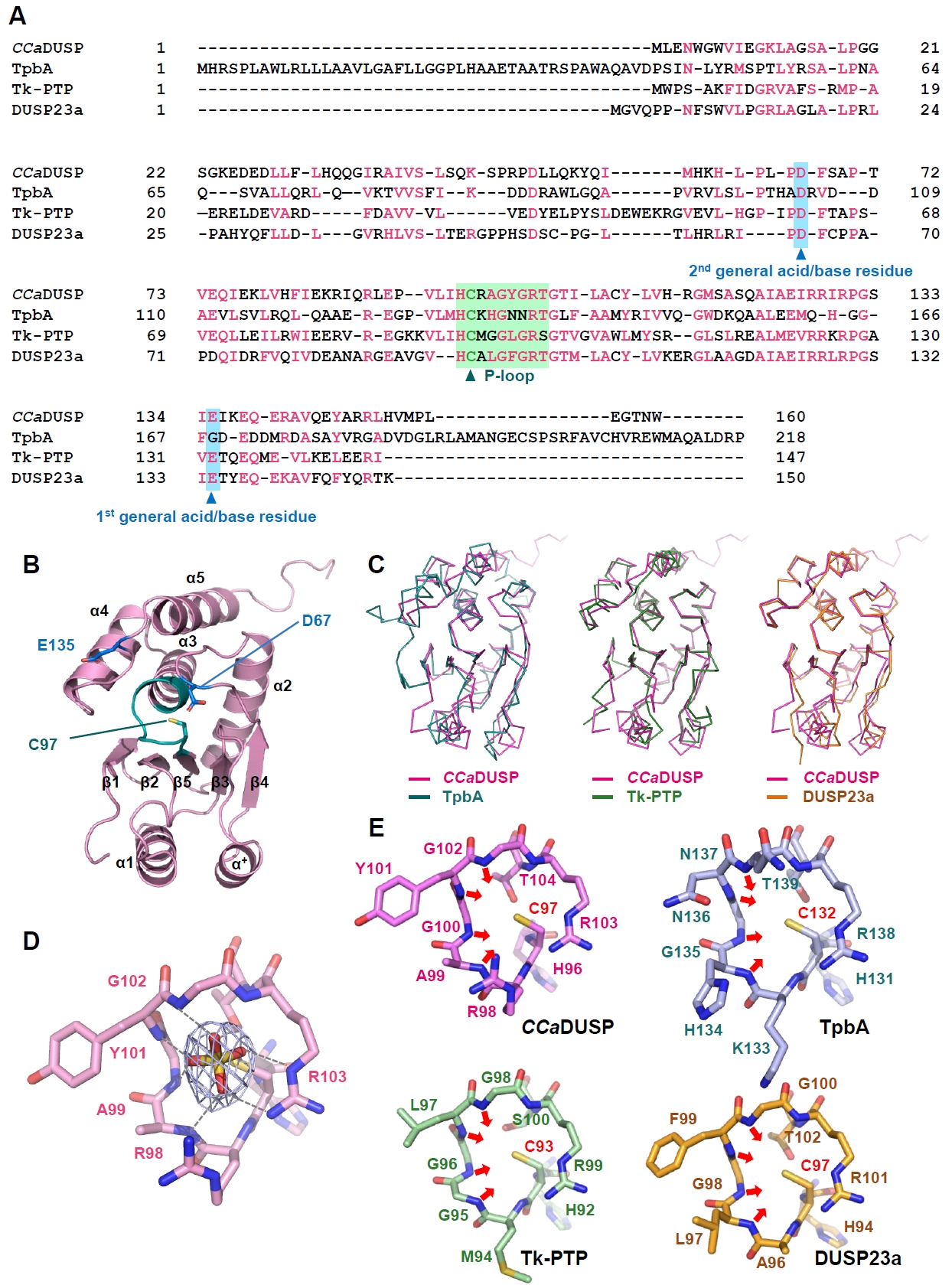
The crystal structure of CCaDUSP was determined to 2.4 Å resolution (Table 2). In the CCaDUSP crystal structure, a central β-sheet is composed of five β-strands [β1(Gly6–Ile9), β2(Lys12–Ser15), β3(Ala39–Ser42), β4(Met59–Leu63), and β5(Val93–His96)], which is sandwiched by six helices: four helices [α2(Thr72–Arg89), α3(Tyr101–Arg115), α4(Ala119–Ile129), and α5(Lys137–His151)] on one side and two helices [α1(Glu25–Gln35) and α+(Pro50–Tyr56)] on the other (Fig. 1B). The overall structure adopts the conventional DUSP fold, composed of a core region (β1–β5 and α2–α5) together with mid-domain variations (α1 and α+) as defined by the DUSP secondary-structure nomenclature (Fig. 1B). Structural superposition showed that CCaDUSP superimposes well with P. aeruginosa TpbA (the sole identified bacterial DUSP before this study), T. kodakarensis Tk-PTP (an archaeal DUSP), and human DUSP23a, with root mean square deviation (RMSD) values of 2.0 Å (over 142 aligned residues), 2.6 Å (over 145 aligned residues), and 1.2 Å (over 137 aligned residues), respectively (Fig. 1C). These results further support that CCaDUSP adopts a canonical DUSP fold.
Structural analysis of the P-loop in CCaDUSP
DUSPs commonly possess a conserved catalytic signature, H−C−x−x−G−x−x−R−S/T, known as the phosphate-binding or P-loop motif (Tabernero et al., 2008; Tautz et al., 2013). In CCaDUSP, this motif resides in the β5−α3 loop and is represented by the sequence H−C97−R−A−G−Y−G−R103. Cys97 is present at the position of the catalytic nucleophile, whereas Arg103 is located at the position of the conserved arginine residue that interacts with and stabilizes the phosphate moiety of the substrate. Notably, a sulfate ion was accommodated in the active site of the CCaDUSP crystal structure, presumably as an analogue of the phosphate group of the substrate (Fig. 1D). This sulfate ion is stabilized by multiple hydrogen bonds with the backbone amides of Ala99, Tyr101, Gly102, and Arg103 and by electrostatic interactions with the guanidinium groups of Arg98 and Arg103 (Fig. 1E). The P-loop of CCaDUSP adopts a catalytically active conformation. The backbone amide groups within the loop, together with the side-chain guanidinium group of Arg103, are oriented toward the catalytic pocket, forming a positively charged surface (Fig. 1D). This electropositive environment complements the negatively charged phosphate group of the substrate and contributes to lowering the pKa of Cys97, thereby maintaining it as a reactive thiolate essential for catalysis (Kolmodin and Aqvist, 2001; Zhang, 2002; Zhang et al., 1994). This arrangement of the P-loop is highly homologous to that observed in catalytically active DUSPs, such as TpbA (PDB code 4R0S), Tk-PTP (PDB code 5Z5A), and DUSP23a (PDB code 4ERC) (Fig. 1E). Collectively, structural analyses indicated that CCaDUSP adopts a canonical DUSP fold with a conserved P-loop.
Enzymatic property of CCaDUSP
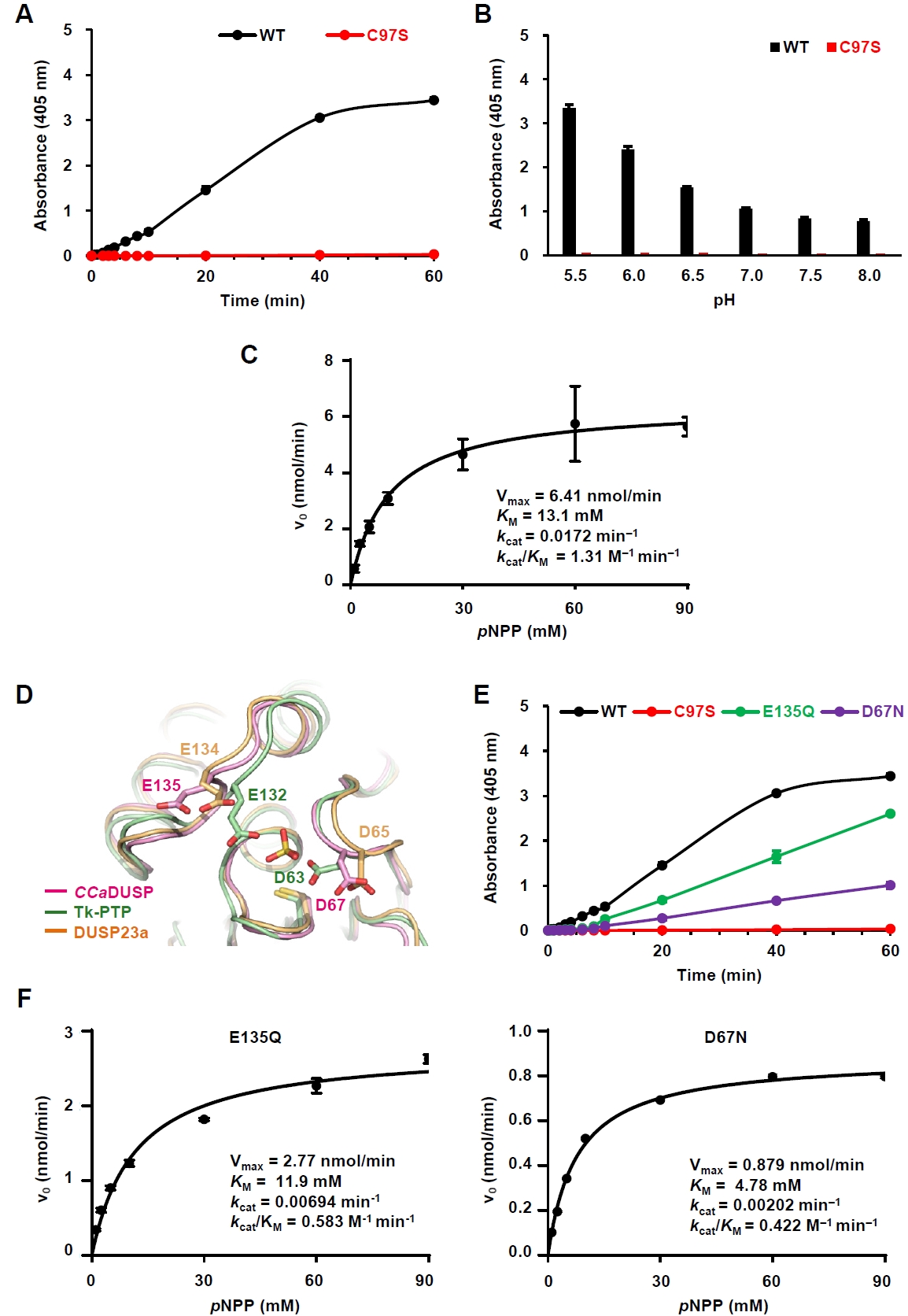
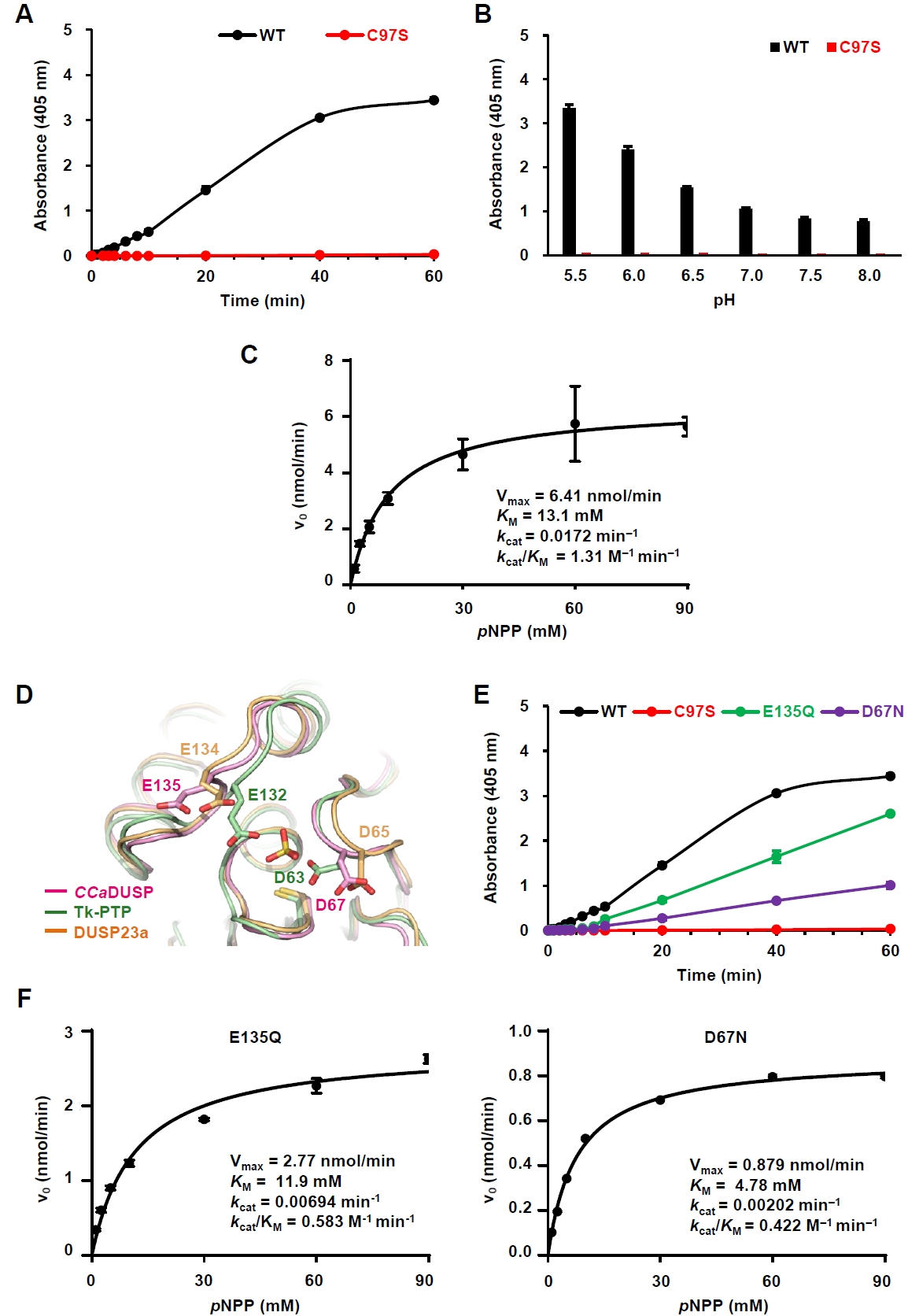
To evaluate the phosphatase activity of CCaDUSP, we performed in vitro biochemical assays using pNPP, a widely used artificial substrate for phosphatases. Wild-type CCaDUSP exhibited significant dephosphorylation activity toward pNPP, whereas the catalytic mutant C97S showed negligible activity (Fig. 2A). Next, we examined the pH dependence of CCaDUSP activity and found that the enzyme exhibited optimal activity at pH 5.5 (Fig. 2B). Notably, C97S mutant displayed only background-level activity. To calculate the kinetic constants of the CCaDUSP dephosphorylation reactions, initial velocity data were obtained and fitted to the Michaelis–Menten equation using nonlinear regression analysis (Fig. 2C). The kcat, KM, and kcat/KM values of CCaDUSP toward pNPP were determined to be 0.0172 min-1, 13.1 mM, and 1.31 M-1 min-1, respectively.
CCaDUSP contains dual general acid/base residues
In addition to the conserved P-loop motif, DUSPs contain another key motif, the D-loop, which includes a conserved aspartate residue that is essential for dephosphorylation. During catalysis, this conserved aspartate residue acts as a general acid/base residue by protonating the leaving group of the substrate phosphate and subsequently activating a water molecule that hydrolyzes the phosphoenzyme intermediate (Tautz et al., 2013). Sequence (Fig. 1A) and structural alignment (Fig. 2D) showed that this aspartate residue is also conserved in CCaDUSP as Asp67, located in the β4−α2 loop. Interestingly, we identified another potential general acid/base residue in CCaDUSP, Glu135, located in the α4−α5 loop. Structural comparison suggested that this residue occupies a position analogous to the secondary general acid/base residues, Glu134 of DUSP23a (Agarwal et al., 2008; Kuznetsov et al., 2012) and Glu132 of Tk−PTP (Yun et al., 2018). To determine whether Asp67 and Glu135 serve as dual acid/base residues in CCaDUSP, we prepared D67N and E135Q mutants and measured their catalytic activities toward pNPP. Both mutations led to a substantial decrease in the dephosphorylation activity compared to that of the wild-type protein (Fig. 2E). Nonlinear regression analysis of the initial velocity data fitted to the Michaelis–Menten equation yielded the catalytic efficiency (kcat/KM) values of 0.583 M-1 min-1 and 0.422 M-1 min-1 for E135Q and D67N mutants, respectively (Fig. 2F). These values are less than half of the kcat/KM of the wild-type protein (1.31 M-1 min-1; Fig. 2C). These results collectively demonstrate that Asp67 and Glu135 cooperate as dual general acid/base residues in the dephosphorylation reaction catalyzed by CCaDUSP.
Conclusion
In this study, we determined the crystal structure of CCaDUSP, which adopts a canonical DUSP fold with a well-conserved catalytic cysteine residue and functions as an active phosphatase. Notably, CCaDUSP represents the second structurally and biochemically characterized bacterial DUSP to date and the first case of a bacterial DUSP harboring dual general acid/base residues. These findings offer insights into the catalytic diversity of bacterial DUSPs and provide a basis for future studies of DUSP family members. Another unresolved question concerns the biological functions of CCaDUSP. Ca. Chx. allophototropha is an anoxygenic phototroph that is likely exposed to fluctuations in light availability and photo-oxidative stress caused by excess light energy (Tsuji et al., 2024). Adaptation to such environmental changes may require the dynamic regulation of protein phosphorylation states, in which CCaDUSP may play a role. Identification of its physiological substrates and interacting partners will contribute to the elucidation of the biological function of CCaDUSP and the significance of its phosphatase activity in the physiology of Ca. Chx. allophototropha.
Acknowledgments
X-ray diffraction data was collected at beamline 5C of the Pohang Accelerator Laboratory, Korea. This work was supported by grants from the National Research Foundation of Korea (RS-2023-00278696), National Research Council of Science and Technology (CRC22021-700), and Korea Research Institute of Bioscience and Biotechnology Research Initiative Programs (KGM9952623 and KQM0052611), supervised by the Ministry of Science and ICT, Korea.
Conflict of Interest
The authors declare that they have no competing interests.
Data Availability
The coordinates of the structure were deposited in the Protein Data Bank with the accession code 25XA, together with the structure factors.
Fig. 1.Sequence and structural analysis of CCaDUSP. (A) The amino acid sequences of CCaDUSP, TpbA, Tk-PTP, and DUSP23a were aligned using Clustal Omega. Conserved residues are highlighted in pink. The phosphate-binding loop (P-loop) and general acid/base residues are shaded in green and sky blue, respectively. The catalytic cysteine and general acid/base aspartate residues are marked with triangles. (B) Crystal structure of CCaDUSP. CCaDUSP is presented as ribbon drawings with secondary structures labeled according to their order of appearance in the primary sequence. CCaDUSP is colored in pink; the P-loop containing the catalytic cysteine residue (Cys97) is in green; dual general acid/base residues are in blue. (C) Structural superposition of CCaDUSP with TpbA (PDB code: 4R0S), Tk-PTP (PDB code: 5Z5A), and DUSP23a (PDB code: 4ERC), which are shown in Cα trace representation. (D) The P-loop of CCaDUSP is shown in sticks. A 2mFo-DFc electron density omit map of the sulfate molecule contoured at 1.6 σ is shown together. Dashed lines indicate electrostatic interactions or hydrogen bonds mediated by the sulfate molecules. (E) The P-loops of four DUSPs are shown as sticks for comparison, as indicated. The direction of the main-chain amides of the four P-loop-constituting central residues is indicated by arrows.
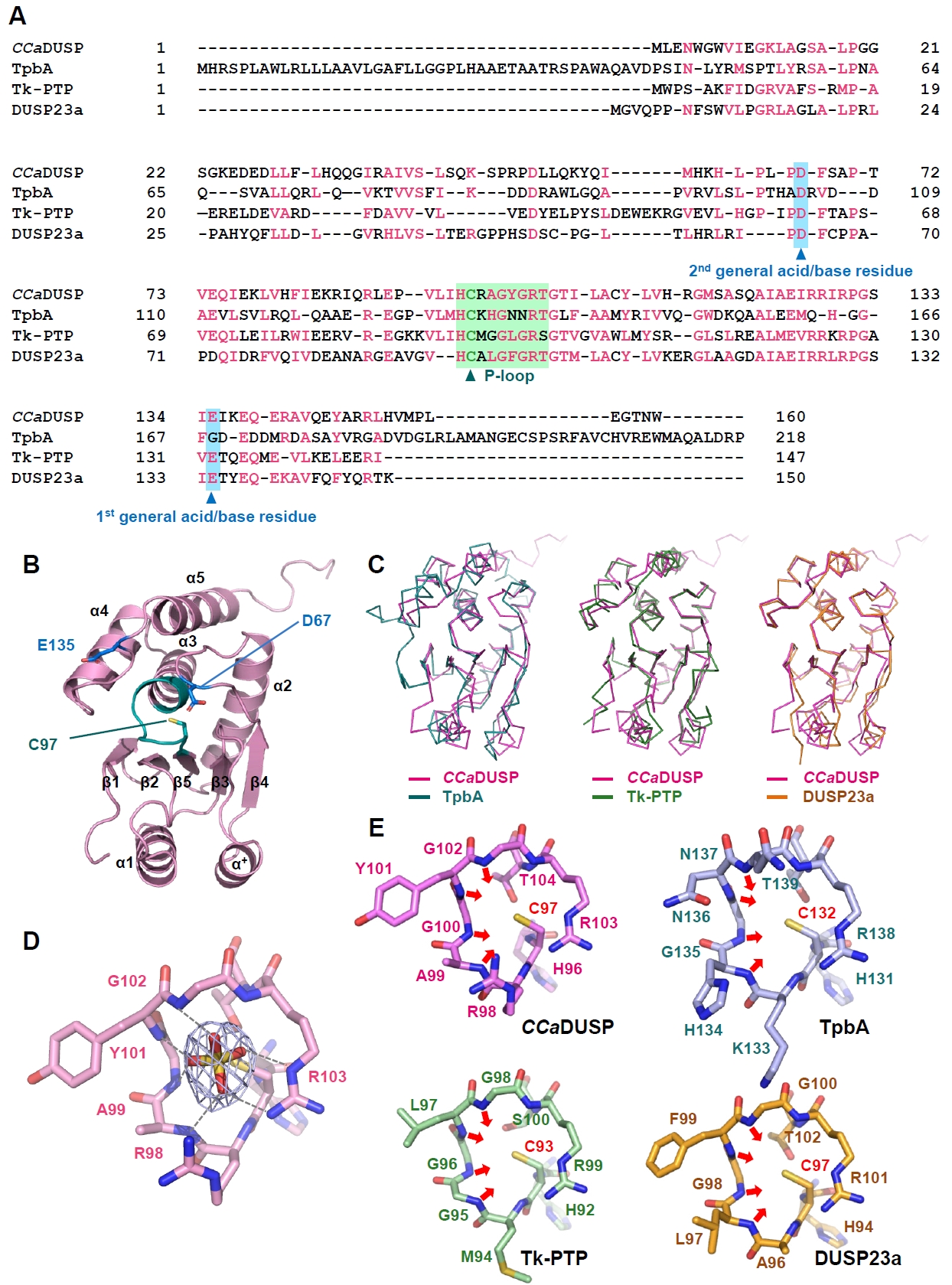
Fig. 2.Characterization of the phosphatase activity of CCaDUSP. All enzymatic assays were performed in triplicate. The details of the experiments are described in the Materials and methods section. (A) Time-dependent hydrolysis of pNPP by wild-type and C97S mutant CCaDUSP was measured at 405 nm over 60 min. (B) Catalytic activity of CCaDUSP was measured 30 min after incubation under the indicated pH conditions using pNPP as a substrate. (C) Michaelis–Menten curves for wild-type CCaDUSP obtained by nonlinear regression analysis using GraphPad Prism. Graphs were derived from initial velocity data collected during the first 5 min of the reaction. The Vmax, KM, kcat, and kcat/KM values are listed. (D) Structural comparison of the positions of the general acid/base residues in CCaDUSP, Tk-PTP, and DUSP23a. (E) Time-dependent pNPP hydrolysis of wild-type and D67N, C97S, or E135Q mutant CCaDUSP proteins. (F) Michaelis–Menten curves for D67N and E135Q mutant CCaDUSP proteins obtained by nonlinear regression analysis using GraphPad Prism. Graphs were derived from initial velocity data collected during the first 5 min of the reaction. The Vmax, KM, kcat, and kcat/KM values for each protein are listed.
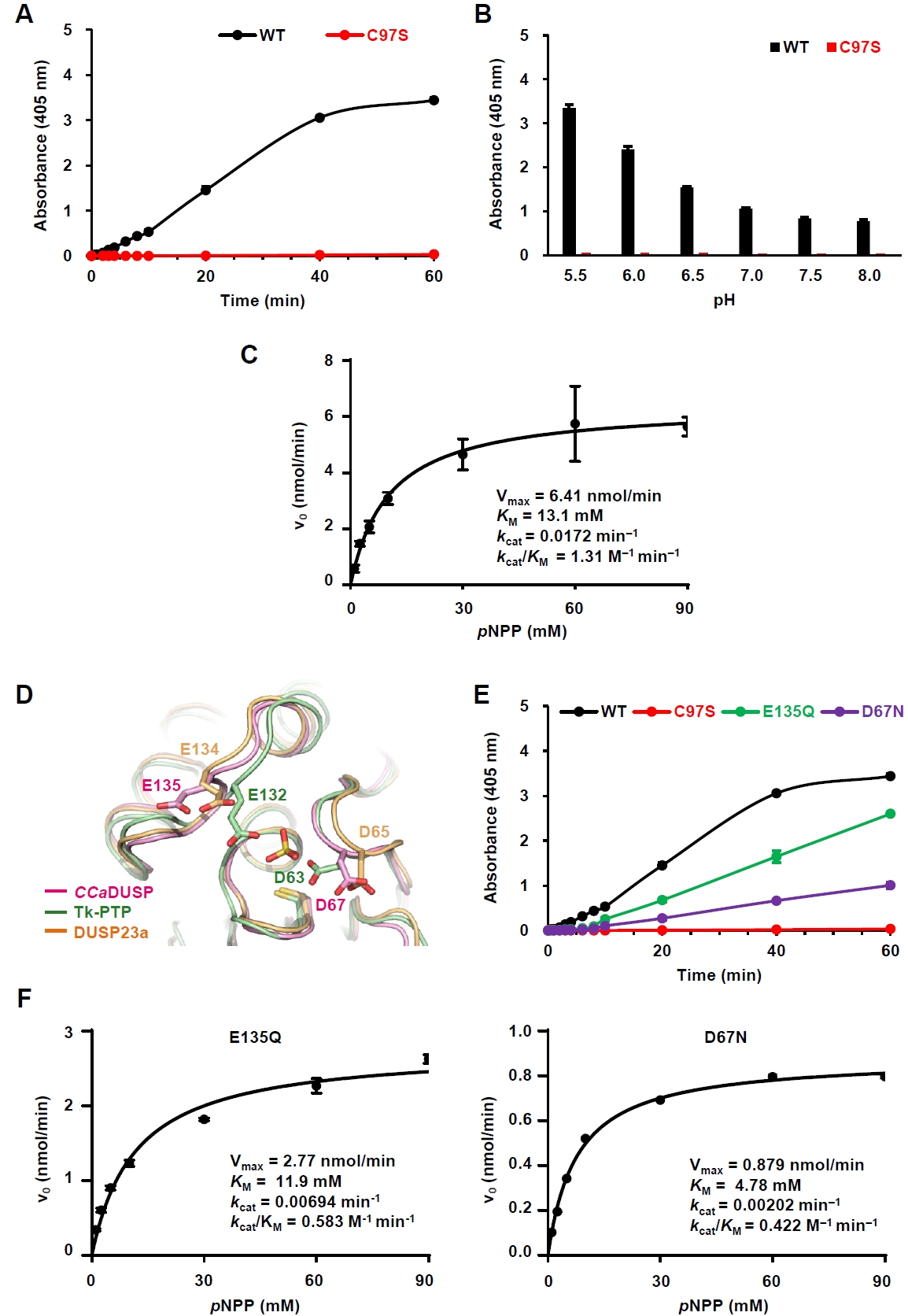
Table 1.Search models used for molecular replacement
|
Search model |
PDB code |
MR solutions |
Top LLG |
Top TFZ |
Space group |
|
AF3-predicted |
- |
1 |
8798.463 |
78.8 |
P63
|
|
TpbA |
4R0S |
19 |
22.705 |
5.6 |
P64
|
|
Tk-PTP |
5Z5A |
1 |
0.425 |
4.8 |
P61
|
|
DUSP23a |
4ERC |
1 |
23.278 |
6.0 |
P63
|
|
DUSP28 |
5Y15 |
30 |
23.634 |
5.6 |
P65
|
Table 2.Data collection and structure refinement statistics
|
Data collection |
PDB code 25XA |
|
Space group |
P63
|
|
Unit cell dimensions |
|
|
a, b, c (Å) |
113.50, 113.50, 66.03 |
|
a, β, γ (°) |
90.00, 90.00, 120.00 |
|
Resolution (Å) |
50.00−2.4 (2.44−2.40)a
|
|
Rsymb
|
0.105 (0.452) |
|
Rmeas
|
0.112 (0.484) |
|
Rpim
|
0.039 (0.170) |
|
CC1/2
|
0.981 (0.896) |
|
CC*
|
0.995 (0.972) |
|
I/σ (I) |
15.7 (2.9) |
|
Completeness (%) |
99.9 (100) |
|
Unique reflections |
19164 (960) |
|
Redundancy |
8.4 (8.5) |
|
Refinement
|
|
|
Resolution (Å) |
50.00−2.40 |
|
Number of reflections |
18983 |
|
Rworkc/Rfree (%) |
0.169/0.214 |
|
Number of atoms |
|
|
Protein |
2551 |
|
Water |
220 |
|
Ligand |
15 |
|
Root-mean-square deviation |
|
|
Bond lengths (Å) |
0.007 |
|
Bond angles (°) |
0.832 |
|
Ramachandran plot (%) |
|
|
Most favored region |
96.85 |
|
Additionally allowed region |
3.15 |
|
Average B-values (Å2) |
|
|
Protein |
37.0 |
|
Water |
41.7 |
|
Ligand |
43.1 |
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, et al. 2010. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 66: 213–221. ArticlePubMedPMC
- Agarwal R, Burley SK, Swaminathan S. 2008. Structure of human dual specificity protein phosphatase 23, VHZ, enzyme-substrate/product complex. J Biol Chem. 283: 8946–8953. ArticlePubMed
- Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, et al. 2004. Protein tyrosine phosphatases in the human genome. Cell. 117: 699–711. ArticlePubMed
- Emsley P, Cowtan K. 2004. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 60: 2126–2132. ArticlePubMed
- Jeong DG, Wei CH, Ku B, Jeon TJ, Chien PN, et al. 2014. The family-wide structure and function of human dual-specificity protein phosphatases. Acta Crystallogr D Biol Crystallogr. 70: 421–435. ArticlePubMed
- Kolmodin K, Aqvist J. 2001. The catalytic mechanism of protein tyrosine phosphatases revisited. FEBS Lett. 498: 208–213. ArticlePubMedLink
- Koveal D, Clarkson MW, Wood TK, Page R, Peti W. 2013. Ligand binding reduces conformational flexibility in the active site of tyrosine phosphatase related to biofilm formation A (TpbA) from Pseudomonas aeruginosa. J Mol Biol. 425: 2219–2231. ArticlePubMedPMC
- Ku B. 2025. Structural analysis of dual specificity phosphatases, the only type of protein tyrosine phosphatases found in humans and across diverse microorganisms. J Microbiol. 63: e2506006. ArticlePubMedPDF
- Ku B, Hong W, Keum CW, Kim M, Ryu H, et al. 2017. Structural and biochemical analysis of atypically low dephosphorylating activity of human dual-specificity phosphatase 28. PLoS One. 12: e0187701. ArticlePubMedPMC
- Kuznetsov VI, Hengge AC, Johnson SJ. 2012. New aspects of the phosphatase VHZ revealed by a high-resolution structure with vanadate and substrate screening. Biochemistry. 51: 9869–9879. ArticlePubMedPMC
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, et al. 2007. Phaser crystallographic software. J Appl Crystallogr. 40: 658–674. ArticlePubMedPMC
- Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276: 307–326. ArticlePubMed
- Patterson KI, Brummer T, O'Brien PM, Daly RJ. 2009. Dual-specificity phosphatases: Critical regulators with diverse cellular targets. Biochem J. 418: 475–489. ArticlePubMedPDF
- Tabernero L, Aricescu AR, Jones EY, Szedlacsek SE. 2008. Protein tyrosine phosphatases: Structure-function relationships. FEBS J. 275: 867–882. ArticlePubMed
- Tautz L, Critton DA, Grotegut S. 2013. Protein tyrosine phosphatases: Structure, function, and implication in human disease. Methods Mol Biol. 1053: 179–221. ArticlePubMedPMC
- Tonks NK. 2006. Protein tyrosine phosphatases: From genes, to function, to disease. Nat Rev Mol Cell Biol. 7: 833–846. ArticlePubMedPDF
- Tsuji JM, Shaw NA, Nagashima S, Venkiteswaran JJ, Schiff SL, et al. 2024. Anoxygenic phototroph of the Chloroflexota uses a type I reaction centre. Nature. 627: 915–922. ArticlePubMedPMCPDF
- Xu K, Li S, Yang W, Li K, Bai Y, et al. 2015. Structural and biochemical analysis of tyrosine phosphatase related to biofilm formation A (TpbA) from the opportunistic pathogen Pseudomonas aeruginosa PAO1. PLoS One. 10: e0124330. ArticlePubMedPMC
- Yun HY, Lee J, Kim H, Ryu H, Shin HC, et al. 2018. Structural study reveals the temperature-dependent conformational flexibility of Tk-PTP, a protein tyrosine phosphatase from Thermococcus kodakaraensis KOD1. PLoS One. 13: e0197635. ArticlePubMedPMC
- Zhang ZY. 2002. Protein tyrosine phosphatases: Structure and function, substrate specificity, and inhibitor development. Annu Rev Pharmacol Toxicol. 42: 209–234. ArticlePubMed
- Zhang ZY, Wang Y, Dixon JE. 1994. Dissecting the catalytic mechanism of protein-tyrosine phosphatases. Proc Natl Acad Sci USA. 91: 1624–1627. ArticlePubMedPMC
Citations
Citations to this article as recorded by


 , Bonsu Ku1,*
, Bonsu Ku1,*
 ePub Link
ePub Link Cite this Article
Cite this Article