- About
- Browse Articles
-
Special Issues
- Pioneering strategies for overcoming bacterial drug resistance (2026)
- Advancing microbial engineering through synthetic biology (2025)
- Host-associated microbiome (2024)
- Bacterial regulatory mechanisms for the control of complex cellular mechanisms (2023)
- Two years into COVID-19 pandemic: Where are we? (2022)
- Collections
- For Contributors
- Policies
- E-Submission

- About
- Browse Articles
-
Special Issues
- Pioneering strategies for overcoming bacterial drug resistance (2026)
- Advancing microbial engineering through synthetic biology (2025)
- Host-associated microbiome (2024)
- Bacterial regulatory mechanisms for the control of complex cellular mechanisms (2023)
- Two years into COVID-19 pandemic: Where are we? (2022)
- Collections
- Policies
- For Contributors
Articles
- Page Path
- HOME > J. Microbiol > Volume 63(7); 2025 > Article
-
Full article
Prebiotic potential of proso millet and quinoa: Effects on gut microbiota composition and functional metabolic pathways - Jinwoo Kim1,2,†, Jiwoon Kim1,†, Yewon Jung1,2, Gyungcheon Kim1, Seongok Kim1,2, Hakdong Shin1,2,*
-
Journal of Microbiology 2025;63(7):e2503002.
DOI: https://doi.org/10.71150/jm.2503002
Published online: July 31, 2025
1Department of Food Science and Biotechnology, College of Life Science, Sejong University, Seoul 05006, Republic of Korea
2Carbohydrate Bioproduct Research Center, Sejong University, Seoul 05006, Republic of Korea
- *Correspondence Hakdong Shin hshin@sejong.ac.kr
- †These authors contributed equally to this work.
© The Author(s), under exclusive licence to Microbiological Society of Korea 2026
This is an Open Access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY 4.0) (https://creativecommons.org/licenses/by/4.0/) which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
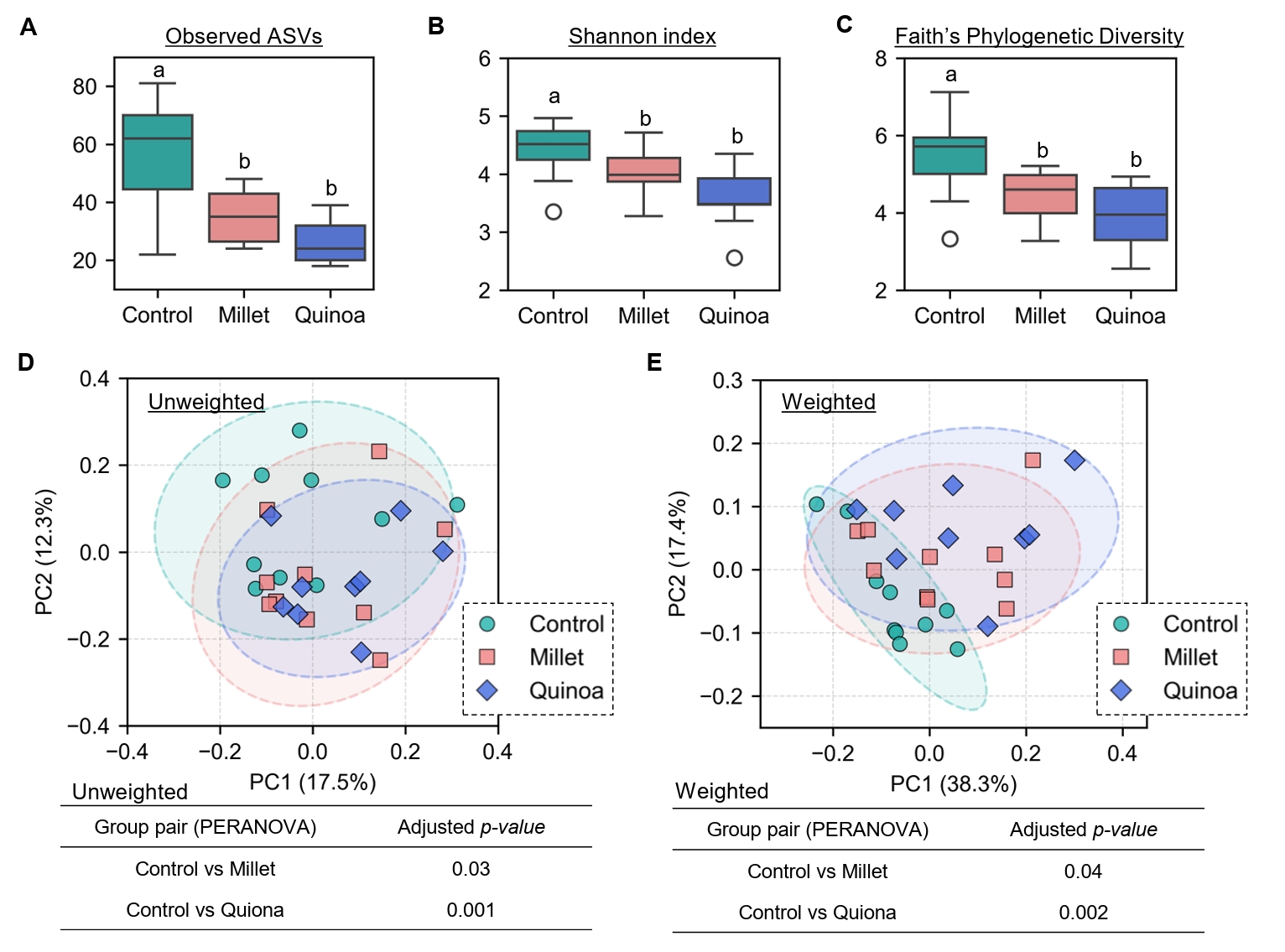
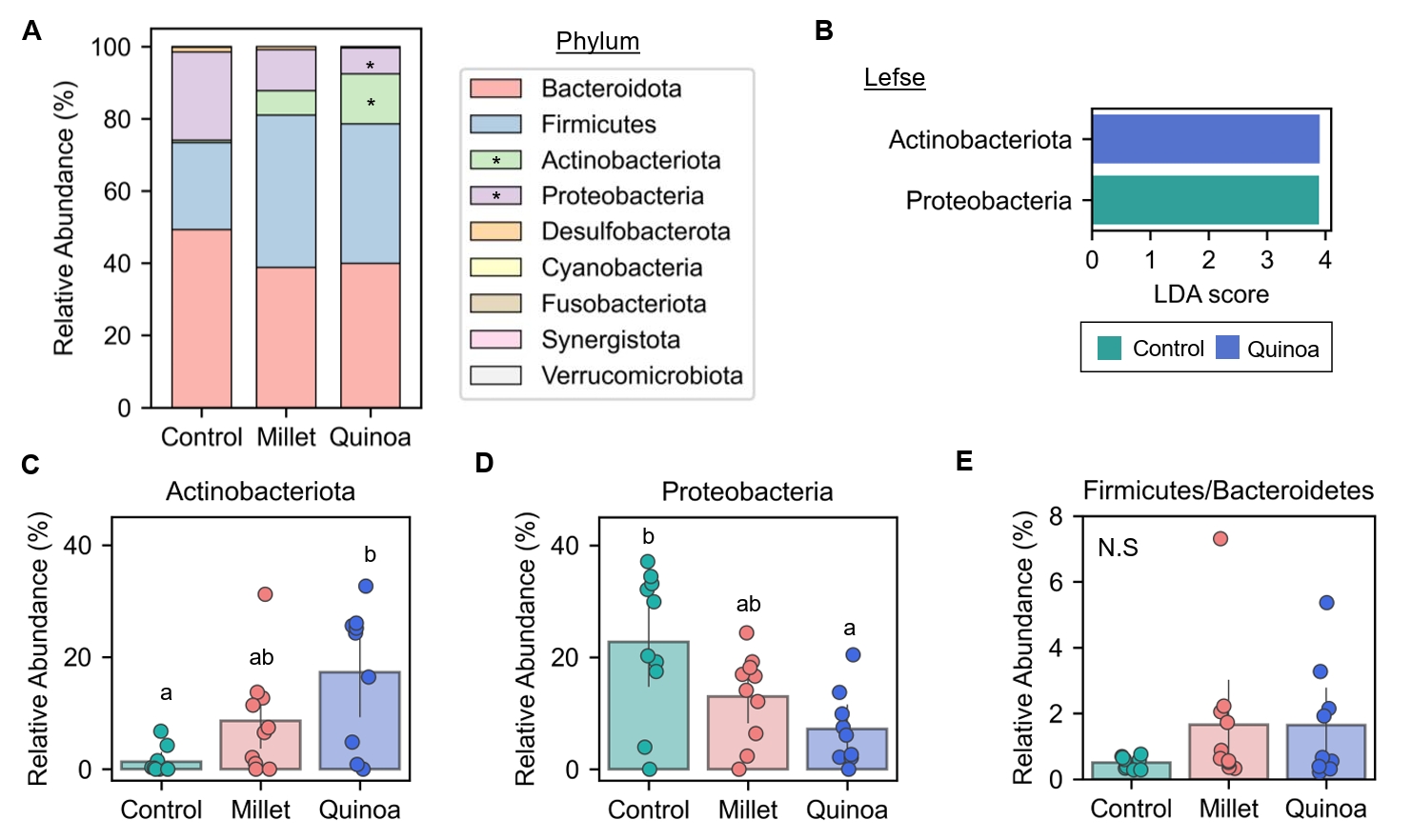
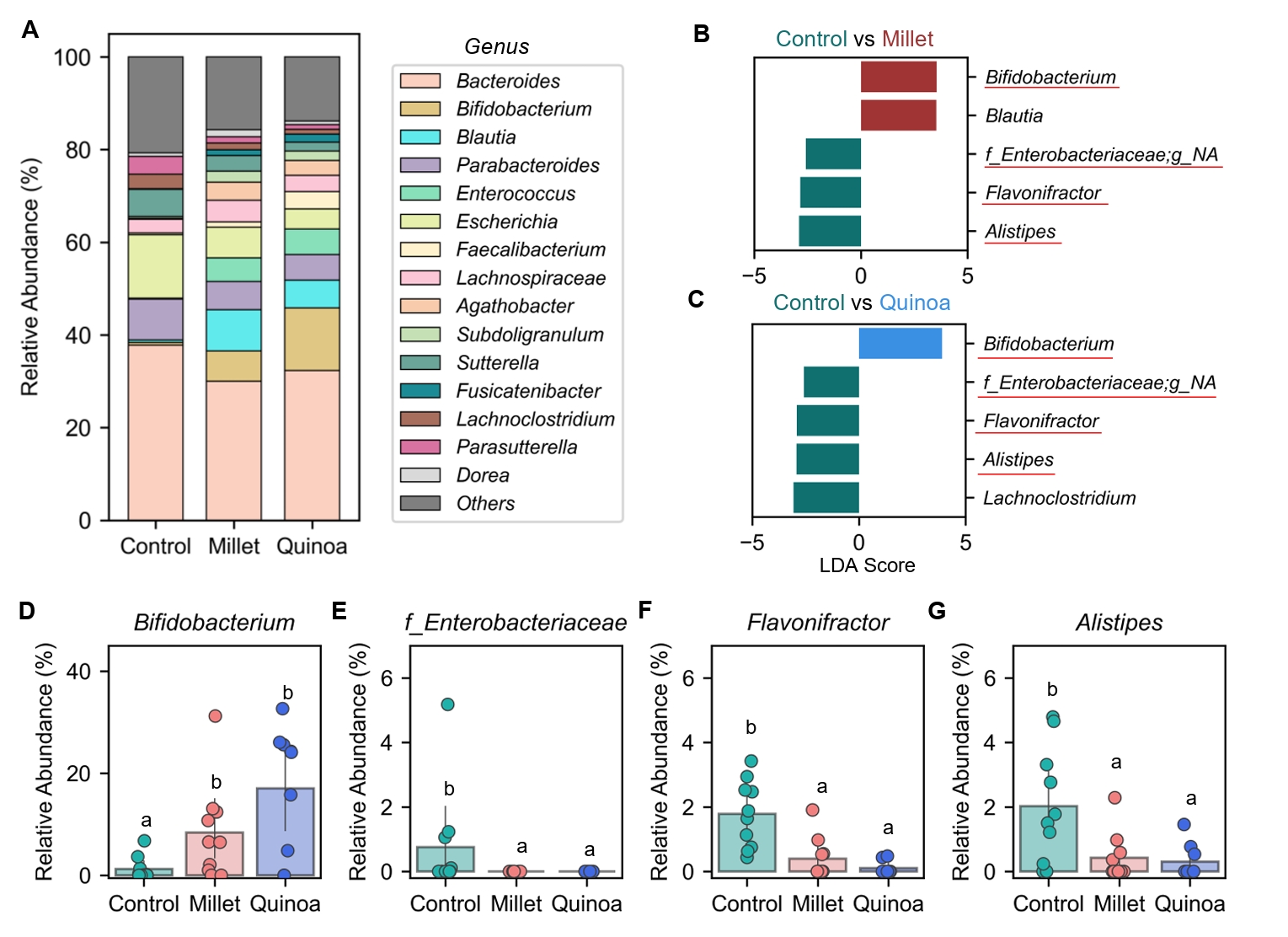
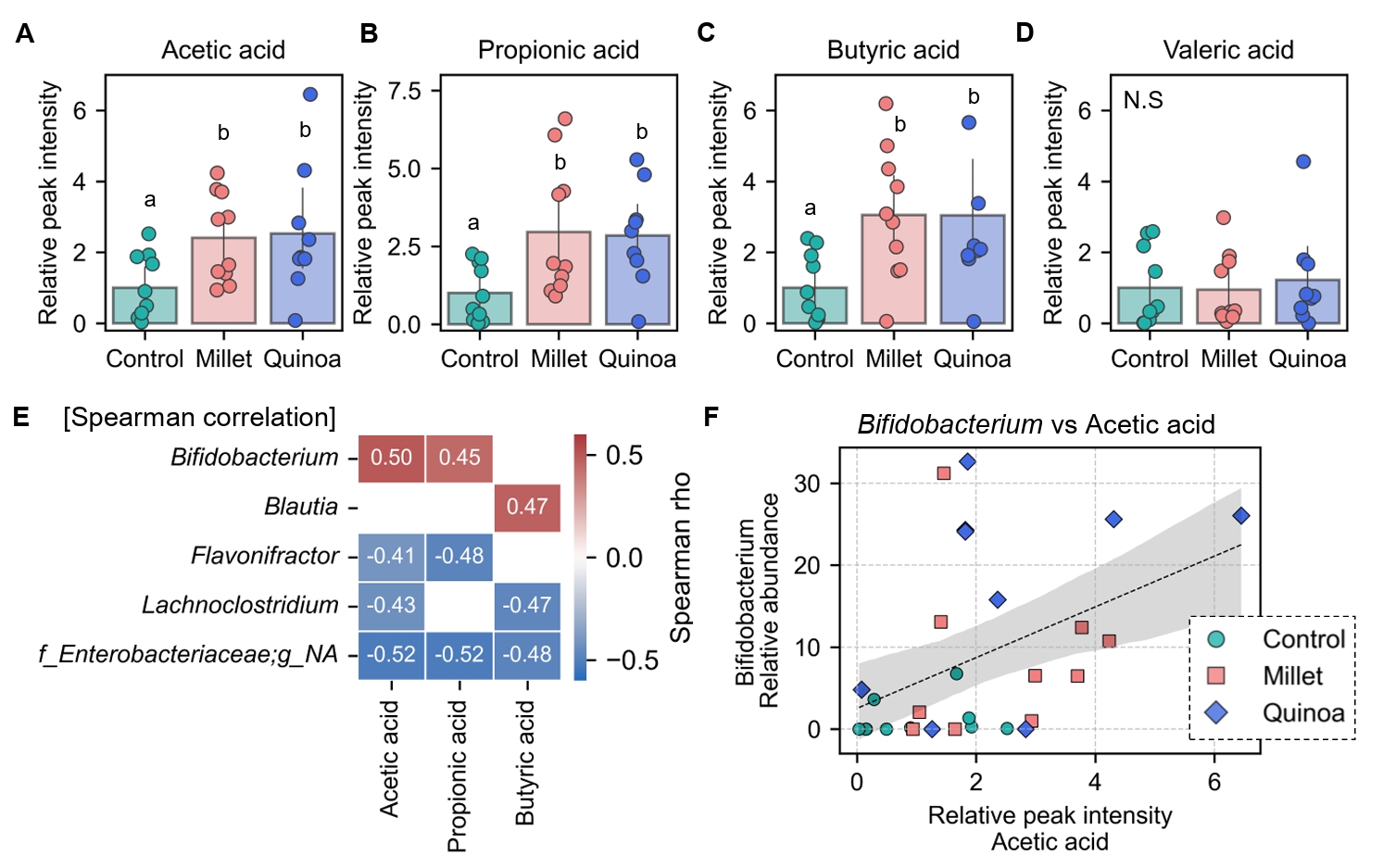
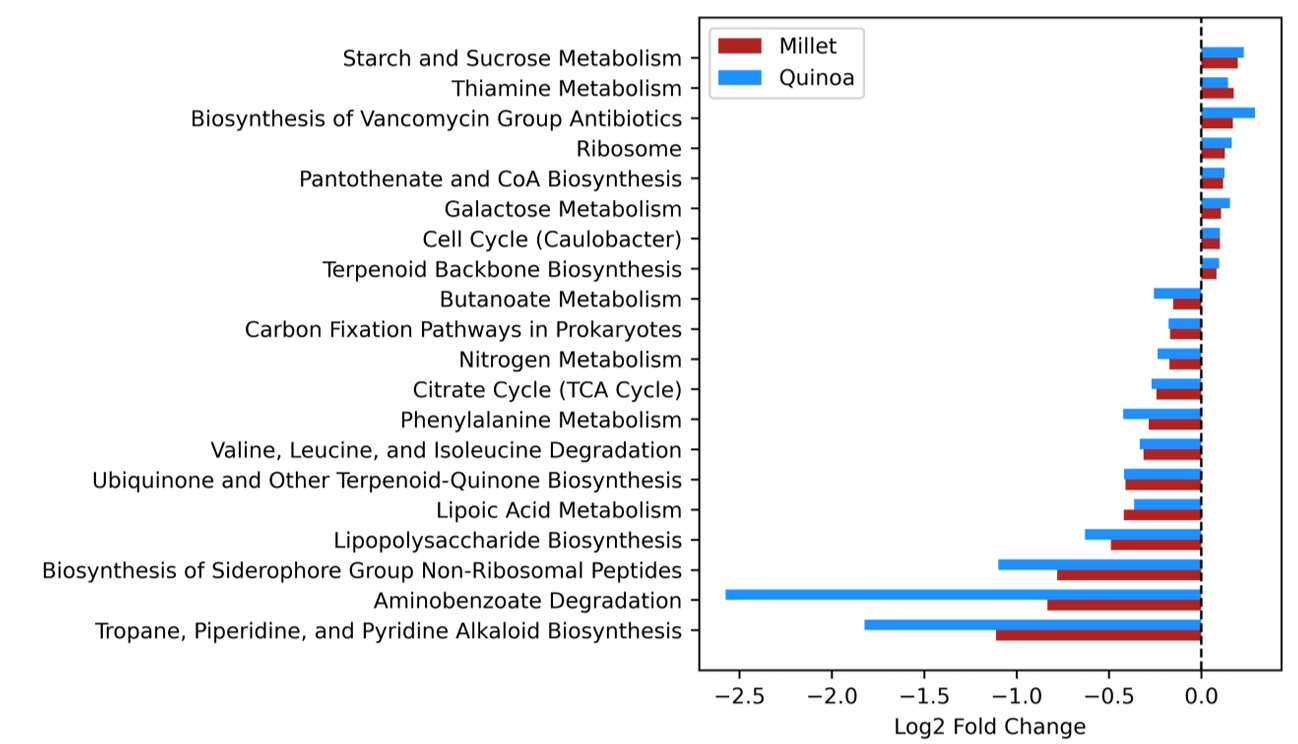
- Prebiotics are indigestible dietary components that improve host health by stimulating the growth and metabolic activity of beneficial intestinal microbes. The whole grains are rich in non-digestible carbohydrates, which may confer prebiotic potential. Among them, millet and quinoa have gained attention as dietary alternatives due to the growing popularity of gluten-free diets. In this study, we examined the effects of proso millet and quinoa on the human gut microbiota using an in vitro fecal incubation model. Both grains altered alpha diversity metrics, including microbial richness, evenness, and phylogenetic diversity. Beta diversity analysis showed that the proso millet and quinoa treatment groups exhibited distinct clustering patterns compared to the control, highlighting their impact on microbial community structure. Taxonomic analysis showed an increase in beneficial genera, including Bifidobacterium, and a decrease in taxa such as Enterobacteriaceae and Flavonifractor. To assess metabolic changes associated with microbial fermentation, short-chain fatty acid (SCFA) intensities were measured. The intensities of acetic acid, propionic acid, and butyric acid were significantly higher in the proso millet- and quinoa-treated groups compared to the control group. Spearman correlation analysis showed that the abundances of Bifidobacterium and Blautia were significantly positively associated with SCFA intensities. Furthermore, predicted functional pathway analysis identified enrichment of carbohydrate-related pathways in proso millet and quinoa treatments. Quinoa supplementation led to a broader enhancement of metabolic pathways, including glycolysis/gluconeogenesis, starch and sucrose metabolism, and pentose phosphate pathways, whereas proso millet enriched galactose metabolism, and starch and sucrose metabolism. These findings suggest that proso millet and quinoa influence gut microbial diversity, composition, and function.
Introduction
Materials and Methods
Results
Discussion
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2022R1A6A1A03055869). This research was also supported by Korea Basic Science Institute grant and Basic Science Research Program funded by the Ministry of Education (No. 2023R1A6C101A045).
Conflict of Interest
The authors declare no conflicts of interest.
Ethical Statements
This study was approved by the Sejong University Institutional Review Board (IRB no. SJU-BR-E-2020-025). Participation was voluntary, and written informed consent was obtained from all participants. The participants were Korean individuals who had not been prescribed antibiotics in the month prior to the experiment and were free from chronic conditions.
Supplementary Information
Table S2.
Table S3.
Fig. S1.
- Aune D, Keum N, Giovannucci E, Fadnes LT, Boffetta P, et al. 2016. Whole grain consumption and risk of cardiovascular disease, cancer, and all cause and cause specific mortality: systematic review and dose-response meta-analysis of prospective studies. BMJ. 353: i2716.ArticlePubMedPMC
- Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, et al. 2019. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 37: 852–857. ArticlePubMedPMC
- Borrello K, Lim U, Park S-Y, Monroe KR, Maskarinec G, et al. 2022. Dietary intake mediates ethnic differences in gut microbial composition. Nutrients. 14: 660.ArticlePubMedPMC
- Brodkorb A, Egger L, Alminger M, Alvito P, Assunção R, et al. 2019. INFOGEST static in vitro simulation of gastrointestinal food digestion. Nat Protoc. 14: 991–1014. ArticlePubMedPDF
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, et al. 2016. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 13: 581–583. ArticlePubMedPMCPDF
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, et al. 2012. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6: 1621–1624. ArticlePubMedPMCPDF
- Chen Y, Zhang R, Xu J, Ren Q. 2022. Alteration of intestinal microflora by the intake of millet porridge improves gastrointestinal motility. Front Nutr. 9: 965687.ArticlePubMedPMC
- Coello K, Hansen TH, Sørensen N, Ottesen NM, Miskowiak KW, et al. 2021. Affective disorders impact prevalence of Flavonifractor and abundance of Christensenellaceae in gut microbiota. Prog Neuropsychopharmacol Biol Psychiatry. 110: 110300.ArticlePubMed
- Cosme F, Inês A, Vilela A. 2022. Consumer’s acceptability and health consciousness of probiotic and prebiotic of non-dairy products. Food Res Int. 151: 110842.ArticlePubMed
- D’Aimmo MR, Satti M, Scarafile D, Modesto M, Pascarelli S, et al. 2023. Folate-producing bifidobacteria: metabolism, genetics, and relevance. Microbiome Res Rep. 3: 11.ArticlePubMedPMC
- de Paulo Farias D, de Araújo FF, Neri-Numa IA, Pastore GM. 2019. Prebiotics: Trends in food, health and technological applications. Trends Food Sci Technol. 93: 23–35. Article
- Eicher TP, Mohajeri MH. 2022. Overlapping mechanisms of action of brain-active bacteria and bacterial metabolites in the pathogenesis of common brain diseases. Nutrients. 14: 2661.ArticlePubMedPMC
- Engevik MA, Morra CN, Röth D, Engevik K, Spinler JK, et al. 2019. Microbial metabolic capacity for intestinal folate production and modulation of host folate receptors. Front Microbiol. 10: 2305.ArticlePubMedPMC
- Esgalhado M, Kemp JA, Damasceno NR, Fouque D, Mafra D. 2017. Short-chain fatty acids: a link between prebiotics and microbiota in chronic kidney disease. Future Microbiol. 12: 1413–1425. Article
- Fu J, Li G, Li X, Song S, Cheng L, et al. 2024. Gut commensal Alistipes as a potential pathogenic factor in colorectal cancer. Discov Oncol. 15: 473.ArticlePubMedPMCPDF
- Fusco W, Lorenzo MB, Cintoni M, Porcari S, Rinninella E, et al. 2023. Short-chain fatty-acid-producing bacteria: Key components of the human gut microbiota. Nutrients. 15: 2211.ArticlePubMedPMC
- Gibson GR, Hutkins R, Sanders ME, Prescott SL, Reimer RA, et al. 2017. Expert consensus document: The International Scientific Association for Probiotics and Prebiotics (ISAPP) consensus statement on the definition and scope of prebiotics. Nat Rev Gastroenterol Hepatol. 14: 491–502. ArticlePDF
- Gullón B, Gullón P, Tavaria FK, Yáñez R. 2016. Assessment of the prebiotic effect of quinoa and amaranth in the human intestinal ecosystem. Food Funct. 7: 3782–3788. Article
- Holmberg SM, Feeney RH, Prasoodanan PKV, Puértolas-Balint F, Singh DK, et al. 2024. The gut commensal Blautia maintains colonic mucus function under low-fiber consumption through secretion of short-chain fatty acids. Nat Commun. 15: 3502.ArticlePMCPDF
- Holscher HD. 2017. Dietary fiber and prebiotics and the gastrointestinal microbiota. Gut Microbes. 8: 172–184. ArticlePubMedPMCLink
- Horiuchi H, Kamikado K, Aoki R, Suganuma N, Nishijima T, et al. 2020. Bifidobacterium animalis subsp. lactis GCL2505 modulates host energy metabolism via the short-chain fatty acid receptor GPR43. Sci Rep. 10: 4158.ArticlePubMedPMCPDF
- Hütt P, Shchepetova J, Loivukene K, Kullisaar T, Mikelsaar M. 2006. Antagonistic activity of probiotic lactobacilli and bifidobacteria against entero‐ and uropathogens. J Appl Microbiol. 100: 1324–1332. Article
- Janda JM, Abbott SL. 2021. The changing face of the family Enterobacteriaceae (Order: “Enterobacterales”): New members, taxonomic issues, geographic expansion, and new diseases and disease syndromes. Clin Microbiol Rev. 34: e00174–20. ArticlePubMedPMCLink
- Jayawardana SAS, Samarasekera JKRR, Hettiarachchi GHCM, Gooneratne J, Mazumdar SD, et al. 2019. Dietary fibers, starch fractions and nutritional composition of finger millet varieties cultivated in Sri Lanka. J Food Compos Anal. 82: 103249.Article
- Johnstone AM, Kelly J, Ryan S, Romero-Gonzalez R, McKinnon H, et al. 2020. Nondigestible carbohydrates affect metabolic health and gut microbiota in overweight adults after weight loss. J Nutr. 150: 1859–1870. ArticlePubMedPDF
- Joye IJ. 2020. Dietary fibre from whole grains and their benefits on metabolic health. Nutrients. 12: 3045.ArticlePubMedPMC
- Khan J, Gul P, Rashid MT, Li Q, Liu K. 2024. Composition of whole grain dietary fiber and phenolics and their impact on markers of inflammation. Nutrients. 16: 1047.ArticlePubMedPMC
- Krishnamurthy VMR, Wei G, Baird BC, Murtaugh M, Chonchol MB, et al. 2012. High dietary fiber intake is associated with decreased inflammation and all-cause mortality in patients with chronic kidney disease. Kidney Int. 81: 300–306. ArticlePubMed
- Langille MG. 2018. Exploring linkages between taxonomic and functional profiles of the human microbiome. mSystems. 3: e00163–17. ArticlePubMedPMCLink
- Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, et al. 2013. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 31: 814–821. ArticlePubMedPMCPDF
- Lemons JM, Conrad M, Tanes C, Chen J, Friedman ES, et al. 2024. Enterobacteriaceae growth promotion by intestinal acylcarnitines, a biomarker of dysbiosis in inflammatory bowel disease. Cell Mol Gastroenterol Hepatol. 17: 131–148. ArticlePubMed
- Leshem A, Segal E, Elinav E. 2020. The gut microbiome and individual-specific responses to diet. mSystems. 5: e00665–20. ArticleLink
- Li L, Abou-Samra E, Ning Z, Zhang X, Mayne J, et al. 2019. An in vitro model maintaining taxon-specific functional activities of the gut microbiome. Nat Commun. 10: 4146.ArticlePubMedPMCPDF
- Li M, Ma S. 2024. A review of healthy role of dietary fiber in modulating chronic diseases. Food Res Int. 174: 114682.Article
- Li C, Niu Z, Zou M, Liu S, Wang M, et al. 2020. Probiotics, prebiotics, and synbiotics regulate the intestinal microbiota differentially and restore the relative abundance of specific gut microorganisms. J Dairy Sci. 103: 5816–5829. ArticlePubMed
- Li Z, Qing Y, Cui G, Li M, Liu T, et al. 2023. Shotgun metagenomics reveals abnormal short-chain fatty acid-producing bacteria and glucose and lipid metabolism of the gut microbiota in patients with schizophrenia. Schizophr Res. 255: 59–66. ArticlePubMed
- Lu Y, Zhou G, Ewald J, Pang Z, Shiri T, et al. 2023. MicrobiomeAnalyst 2.0: Comprehensive statistical, functional and integrative analysis of microbiome data. Nucleic Acids Res. 51: W310–W318. ArticlePubMedPMCPDF
- Nowak V, Du J, Charrondière UR. 2016. Assessment of the nutritional composition of quinoa (Chenopodium quinoa Willd.). Food Chem. 193: 47–54. ArticlePubMed
- Parker BJ, Wearsch PA, Veloo AC, Rodriguez-Palacios A. 2020. The genus Alistipes: Gut bacteria with emerging implications to inflammation, cancer, and mental health. Front Immunol. 11: 906.ArticlePubMedPMC
- Partula V, Deschasaux M, Druesne-Pecollo N, Latino-Martel P, Desmetz E, et al. 2020. Associations between consumption of dietary fibers and the risk of cardiovascular diseases, cancers, type 2 diabetes, and mortality in the prospective NutriNet-Santé cohort. Am J Clin Nutr. 112: 195–207. ArticlePubMedPDF
- Peng M, Tabashsum Z, Anderson M, Truong A, Houser AK, et al. 2020. Effectiveness of probiotics, prebiotics, and prebiotic‐like components in common functional foods. Compr Rev Food Sci Food Saf. 19: 1908–1933. ArticlePubMedLink
- Price MN, Dehal PS, Arkin AP. 2010. FastTree 2 - approximately maximum-likelihood trees for large alignments. PLoS One. 5: e9490. ArticlePubMedPMC
- Pulvento C, Riccardi M, Lavini A, Iafelice G, Marconi E, et al. 2012. Yield and quality characteristics of quinoa grown in open field under different saline and non‐saline irrigation regimes. J Agron Crop Sci. 198: 254–263. Article
- Quince C, Walker AW, Simpson JT, Loman NJ, Segata N. 2017. Shotgun metagenomics, from sampling to analysis. Nat Biotechnol. 35: 833–844. ArticlePubMedPDF
- Radivojac P, Clark WT, Oron TR, Schnoes AM, Wittkop T, et al. 2013. A large-scale evaluation of computational protein function prediction. Nat Methods. 10: 221–227. ArticlePubMedPMC
- Schöpping M, Gaspar P, Neves AR, Franzén CJ, Zeidan AA. 2021. Identifying the essential nutritional requirements of the probiotic bacteria Bifidobacterium animalis and Bifidobacterium longum through genome-scale modeling. NPJ Syst Biol Appl. 7: 47.ArticlePubMedPMC
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, et al. 2011. Metagenomic biomarker discovery and explanation. Genome Biol. 12: R60.ArticlePubMedPMCPDF
- Slavin JL, Jacobs D, Marquart L. 2000. Grain processing and nutrition. Crit Rev Food Sci Nutr. 40: 309–326. ArticlePubMed
- Su Q, Zhuang D-H, Li Y-C, Chen Y, Wang X-Y, et al. 2024. Gut microbiota contributes to high-altitude hypoxia acclimatization of human populations. Genome Biol. 25: 232.ArticlePubMedPMCPDF
- Usta-Gorgun B, Yilmaz-Ersan L. 2020. Short-chain fatty acids production by Bifidobacterium species in the presence of salep. Electron J Biotechnol. 47: 29–35. Article
- Wang H, Huang X, Tan H, Chen X, Chen C, et al. 2022. Interaction between dietary fiber and Bifidobacteria in promoting intestinal health. Food Chem. 393: 133407.ArticlePubMed
- Woomer JS, Adedeji AA. 2021. Current applications of gluten-free grains - a review. Crit Rev Food Sci Nutr. 61: 14–24. ArticlePubMed
- Xiao M, Zhang C, Duan H, Narbad A, Zhao J, et al. 2024. Cross-feeding of Bifidobacteria promotes intestinal homeostasis: A lifelong perspective on the host health. NPJ Biofilms Microbiomes. 10: 47.ArticlePubMedPMCPDF
- Yilmaz P, Parfrey LW, Yarza P, Gerken J, Pruesse E, et al. 2014. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 42: D643–D648. ArticlePubMed
- Yoshii K, Hosomi K, Sawane K, Kunisawa J. 2019. Metabolism of dietary and microbial vitamin B family in the regulation of host immunity. Front Nutr. 6: 48.ArticlePubMedPMC
- Zeyneb H, Pei H, Cao X, Wang Y, Win Y, et al. 2021. In vitro study of the effect of quinoa and quinoa polysaccharides on human gut microbiota. Food Sci Nutr. 9: 5735–5745. ArticlePubMedPMCLink
- Zhao S, Pan C, Zhao J, Du H, Li M, et al. 2022. Quantitative proteomic analysis of the microbial degradation of 3-aminobenzoic acid by Comamonas sp. QT12. Sci Rep. 12: 17609.ArticlePubMedPMCPDF
- Ziegler K, Braun K, Böckler A, Fuchs G. 1987. Studies on the anaerobic degradation of benzoic acid and 2-aminobenzoic acid by a denitrifying Pseudomonas strain. Arch Microbiol. 149: 62–69. ArticlePDF
References
Supplementary Information
References
Citations
- Red quinoa hydrolysate as a plant-based therapeutic alternative for damage induced by high cadmium concentrations to the vascular system in rats
Samia Hassan Husein Kanaan, Paola Zambelli Moraes, Katye Yasmin de Souza de Oliveira, Fernando Barbosa, José Eudes Gomes Pinheiro, Franck Maciel Peçanha, Dalton Valentim Vassallo, Marta Miguel-Castro, Giulia Alessandra Wiggers
Food & Function.2026; 17(8): 3749. CrossRef - Proso Millet (Panicum miliaceum): Nutritional Composition, Functional Attributes, and Health Implications
Sangeeta Yadav, Pratiksha Singh, Mazia Ahmed, Pinki Saini
Future Postharvest and Food.2026;[Epub] CrossRef
 ePub Link
ePub Link Cite this Article
Cite this Article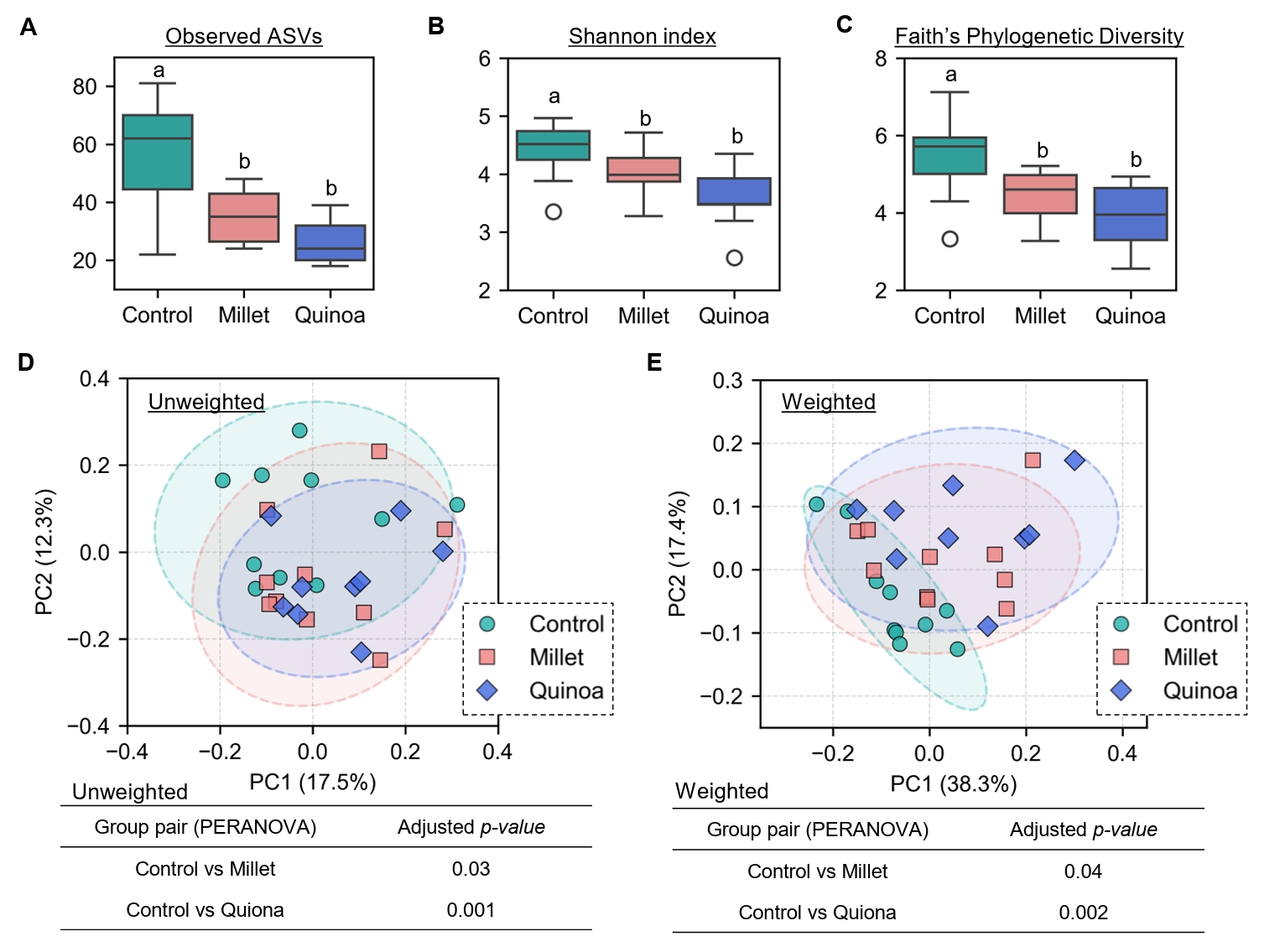
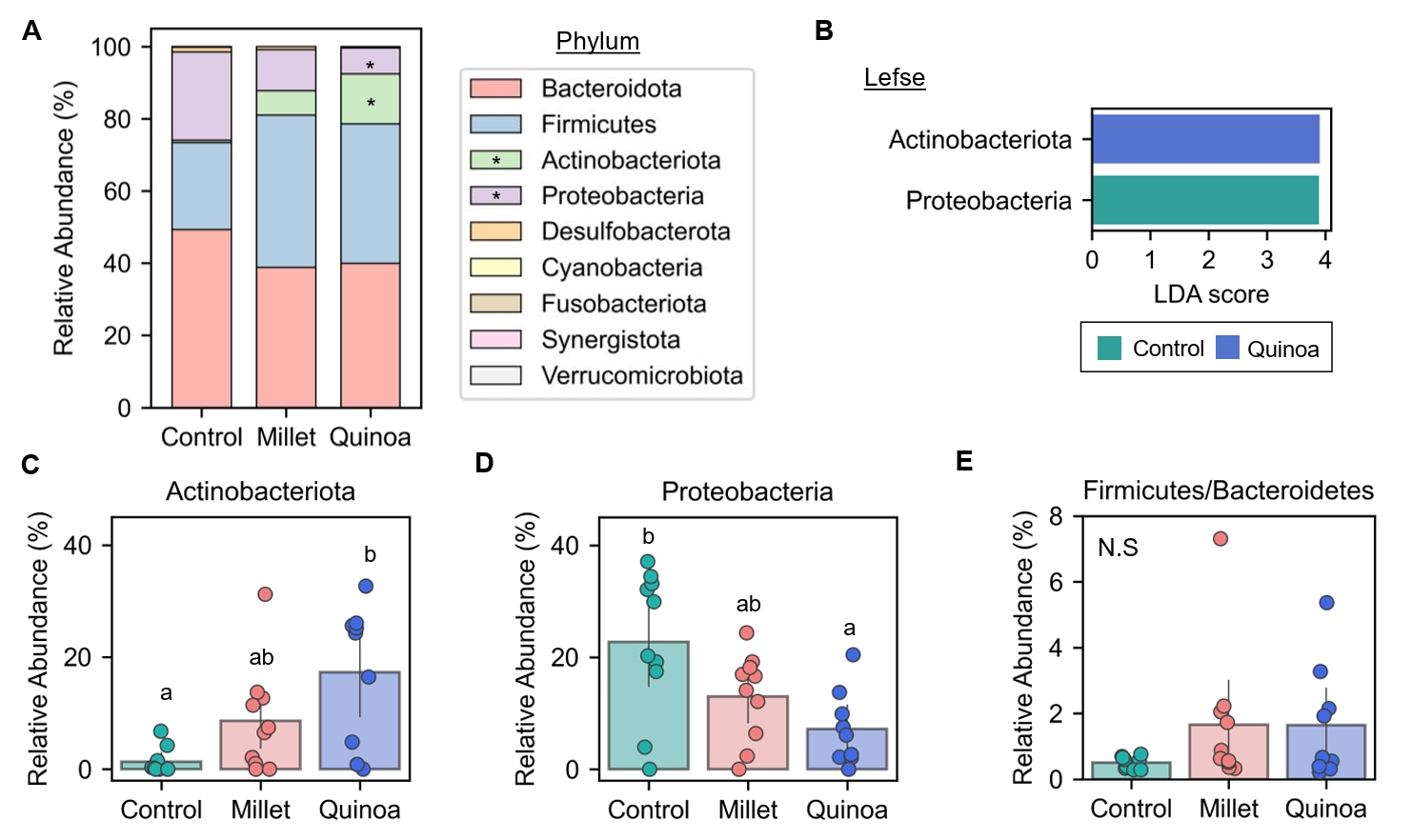
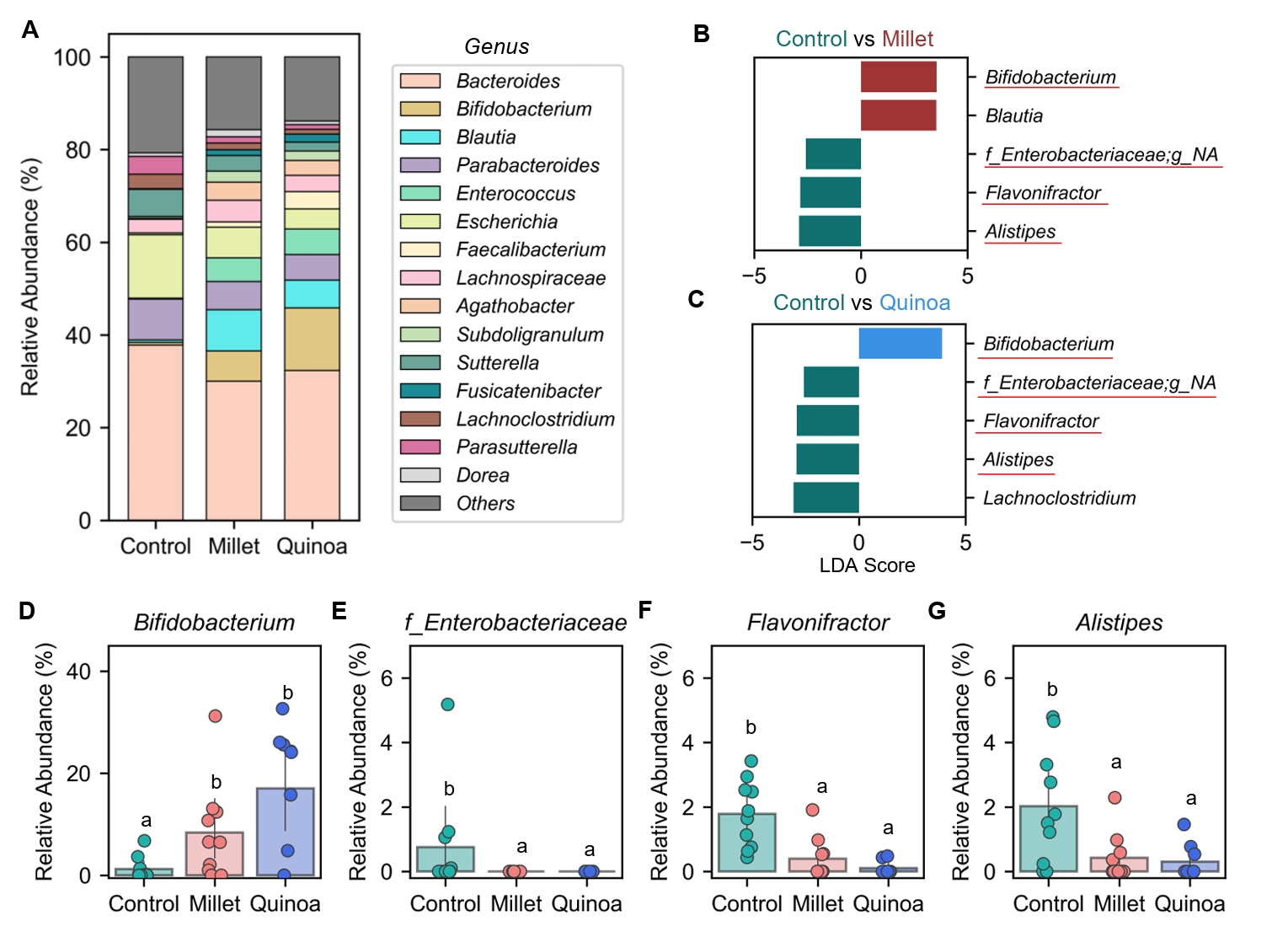
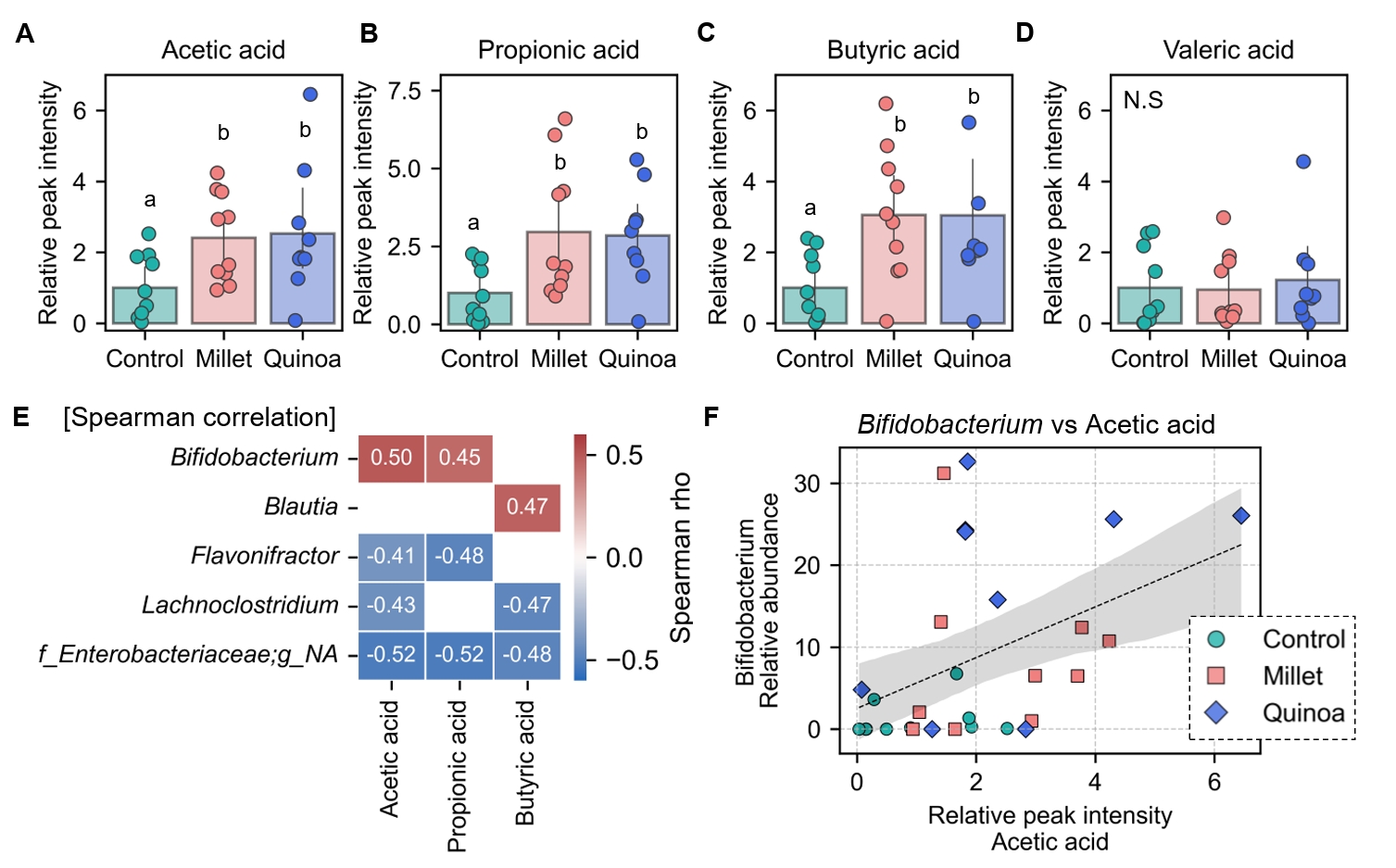
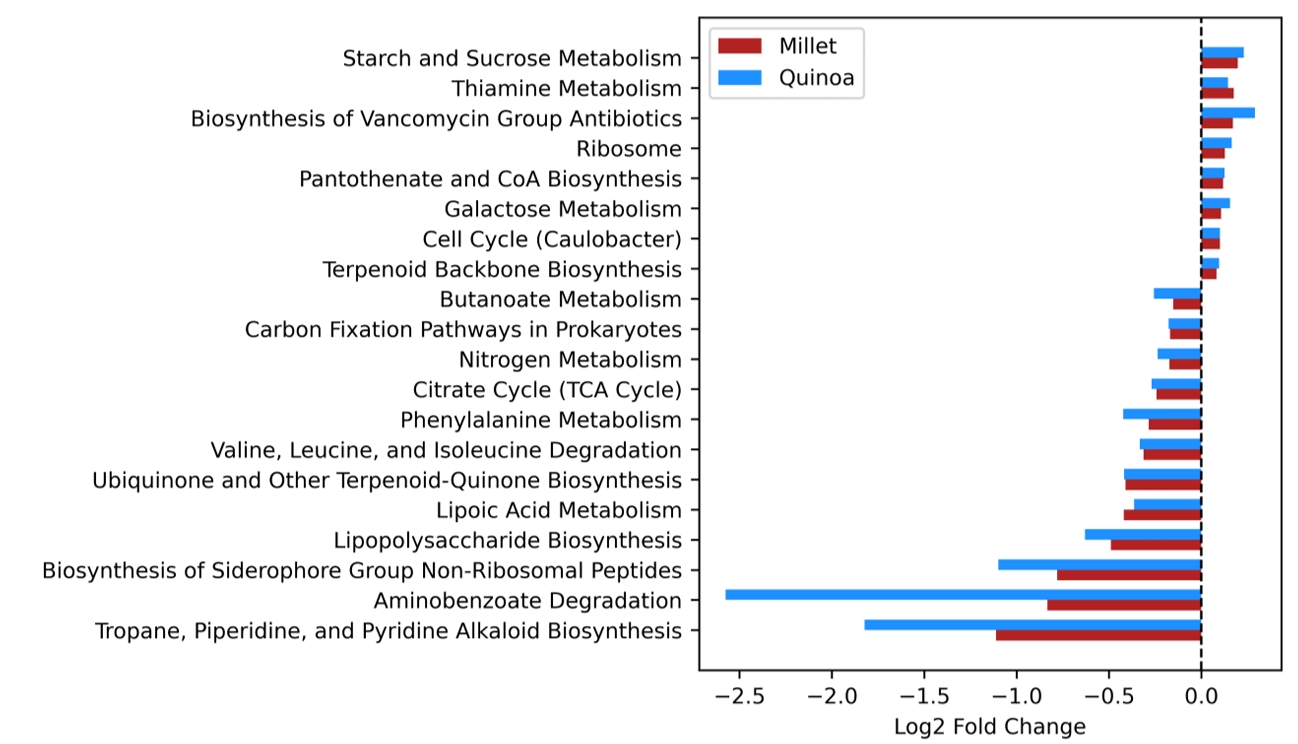
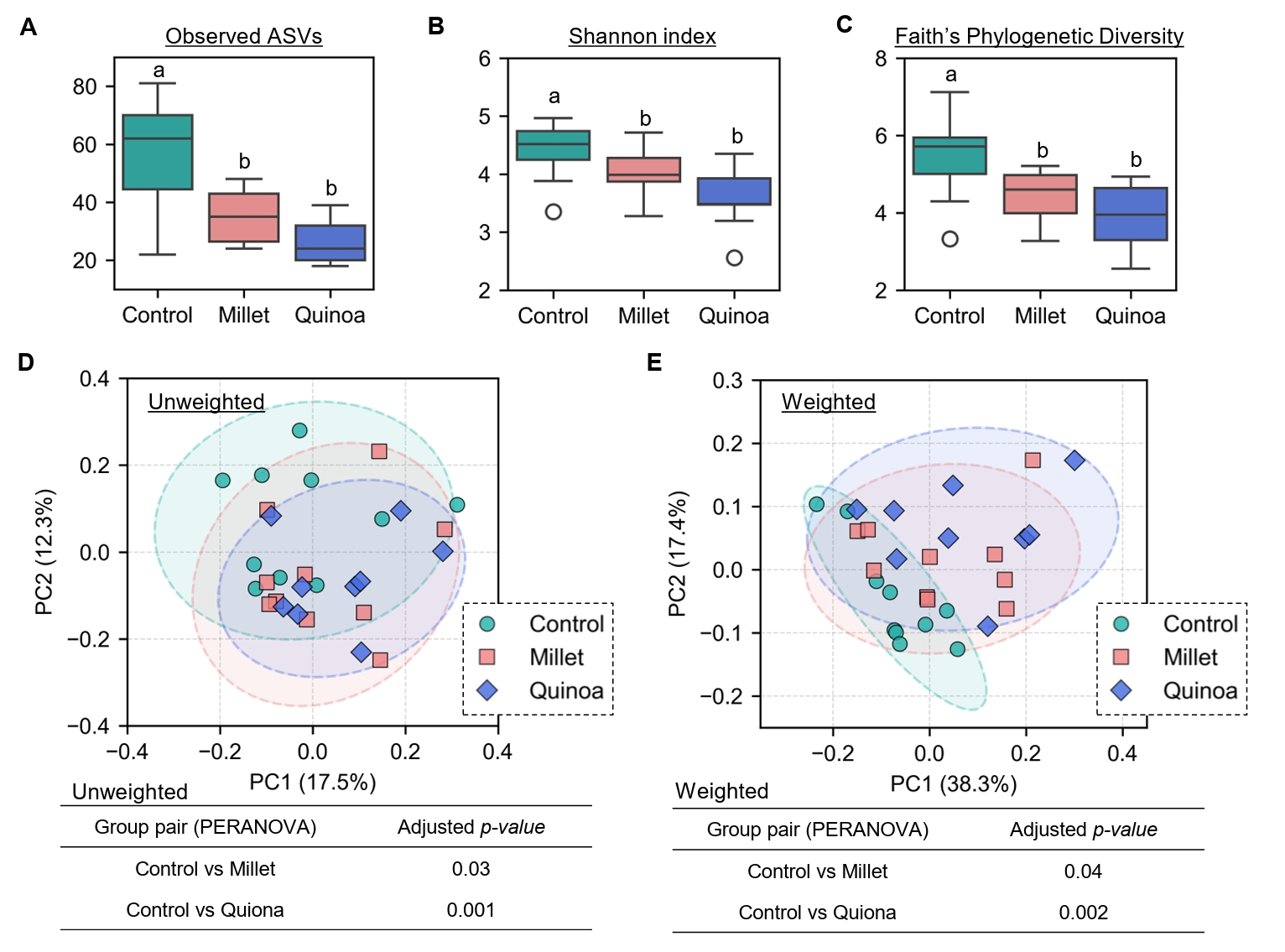
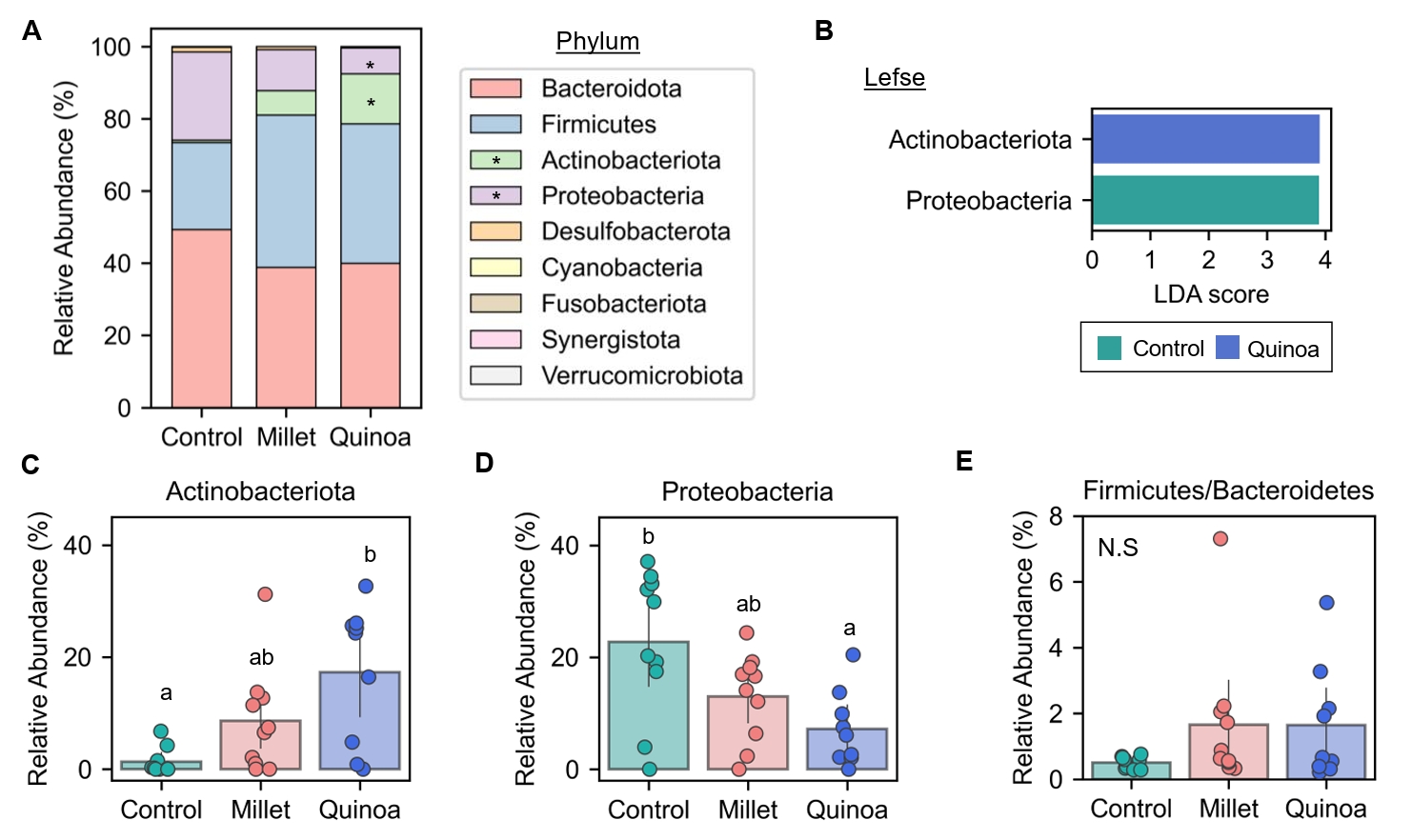
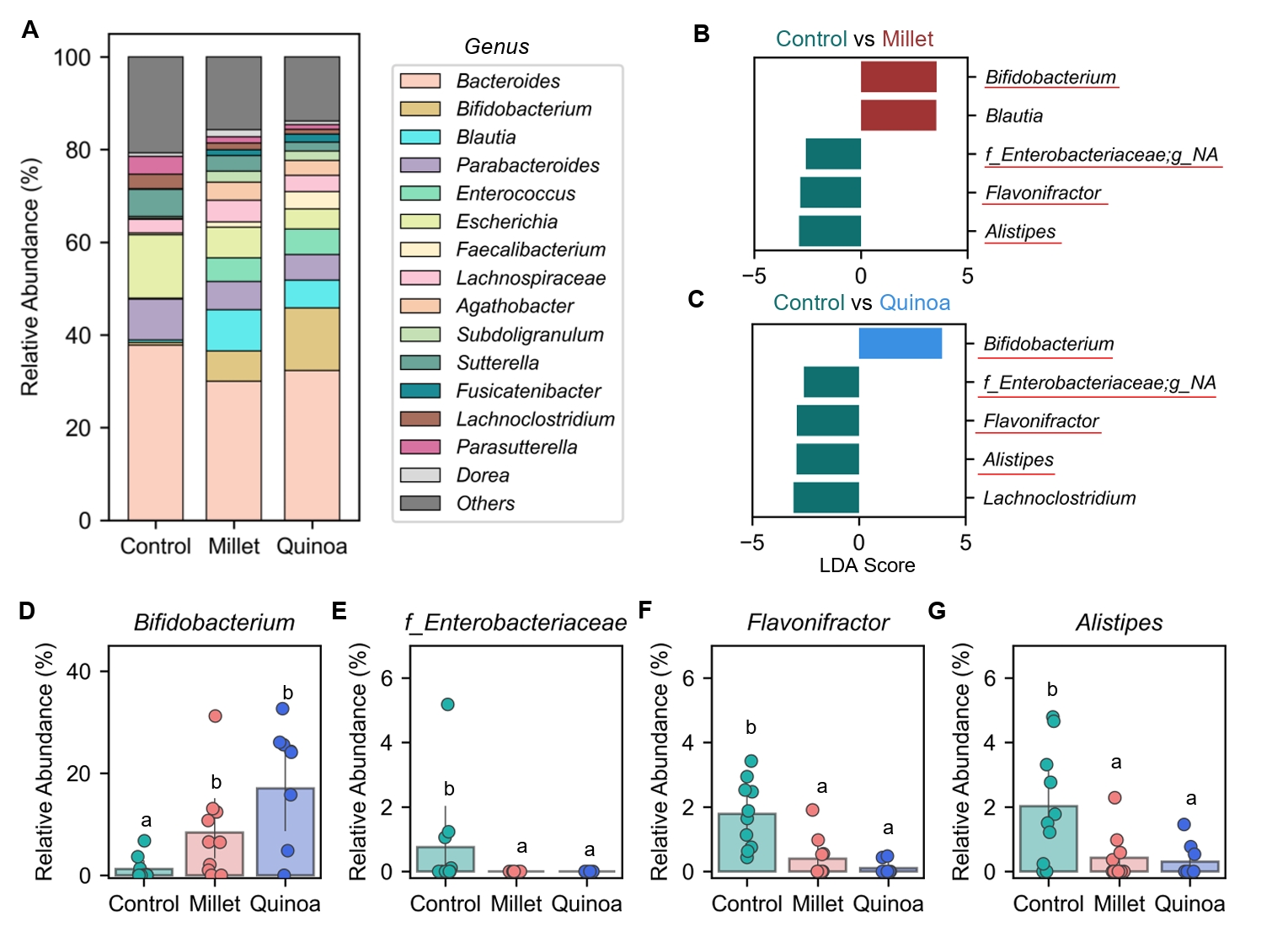
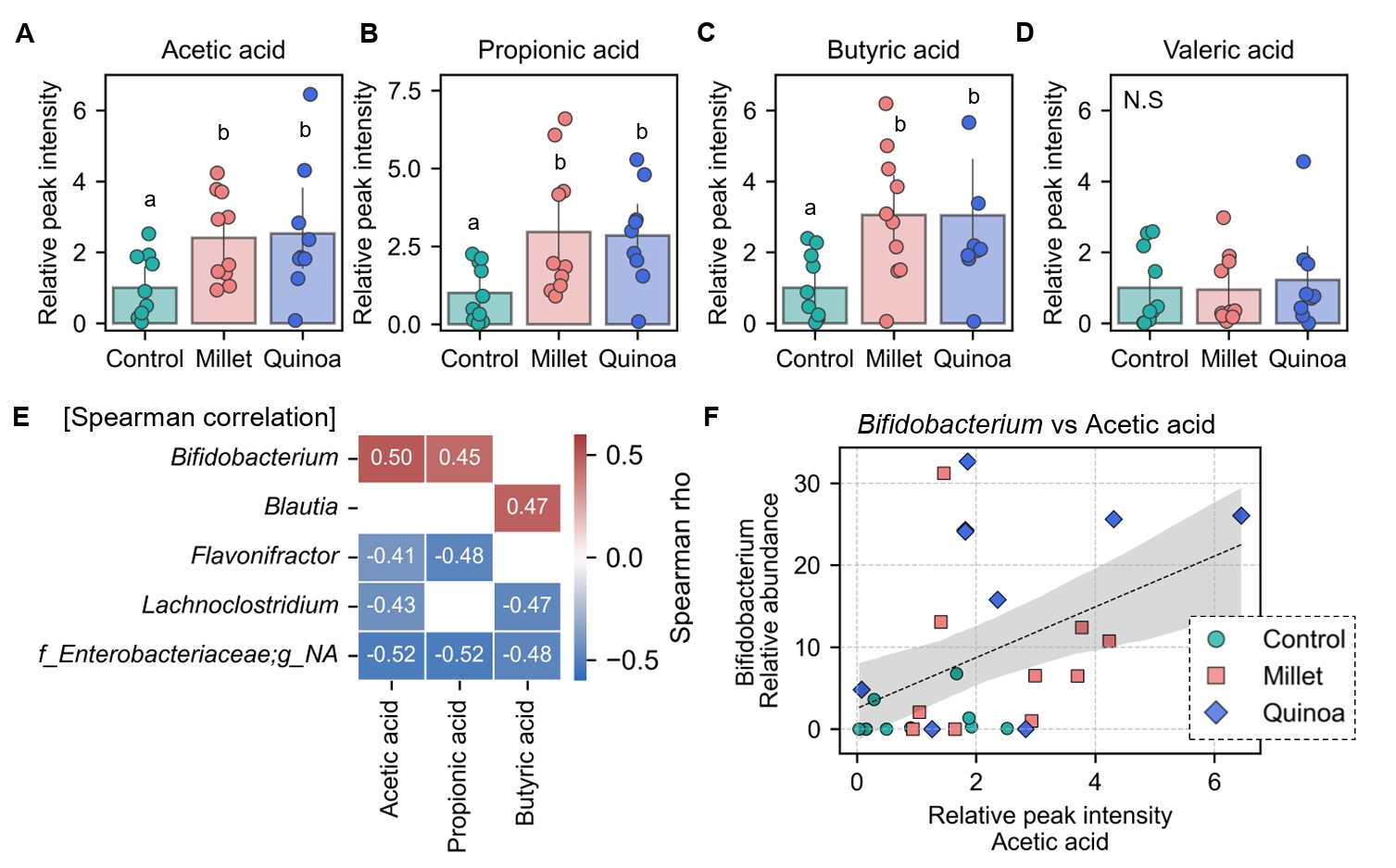
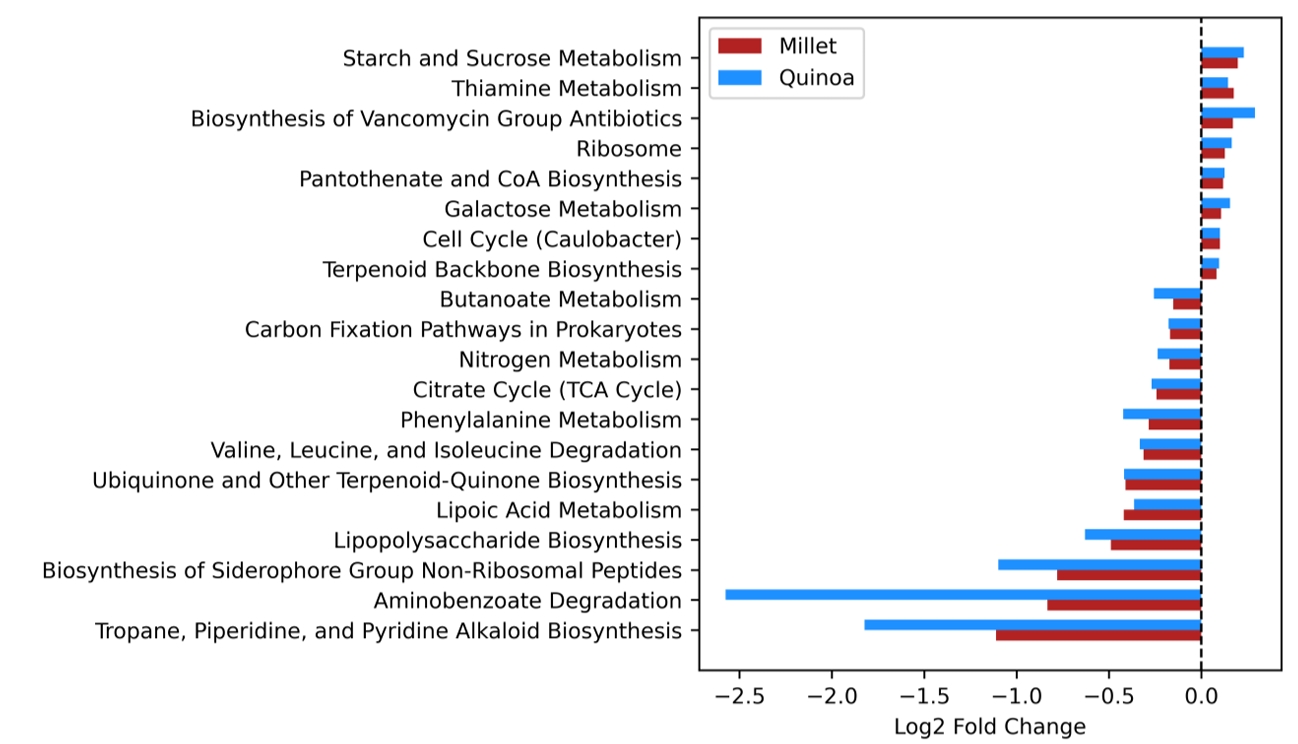
Fig. 1.
Fig. 2.
Fig. 3.
Fig. 4.
Fig. 5.
TOP

