- About
- Browse Articles
-
Special Issues
- Pioneering strategies for overcoming bacterial drug resistance (2026)
- Advancing microbial engineering through synthetic biology (2025)
- Host-associated microbiome (2024)
- Bacterial regulatory mechanisms for the control of complex cellular mechanisms (2023)
- Two years into COVID-19 pandemic: Where are we? (2022)
- Collections
- For Contributors
- Policies
- E-Submission

- About
- Browse Articles
-
Special Issues
- Pioneering strategies for overcoming bacterial drug resistance (2026)
- Advancing microbial engineering through synthetic biology (2025)
- Host-associated microbiome (2024)
- Bacterial regulatory mechanisms for the control of complex cellular mechanisms (2023)
- Two years into COVID-19 pandemic: Where are we? (2022)
- Collections
- Policies
- For Contributors
Articles
- Page Path
- HOME > J. Microbiol > Volume 63(1); 2025 > Review
-
Minireview
Advances in functional analysis of the microbiome: Integrating metabolic modeling, metabolite prediction, and pathway inference with Next-Generation Sequencing data -
Sungwon Jung1,2

-
Journal of Microbiology 2025;63(1):e.2411006.
DOI: https://doi.org/10.71150/jm.2411006
Published online: January 24, 2025
1Department of Genome Medicine and Science, Gachon University College of Medicine, Incheon 21565, Republic of Korea
2Gachon Institute of Genome Medicine and Science, Gachon University Gil Medical Center, Incheon 21565, Republic of Korea
- Sungwon Jung E-mail: sjung@gachon.ac.kr
• Received: November 5, 2024 • Revised: November 22, 2024 • Accepted: November 27, 2024
© The Author(s), under exclusive licence to Microbiological Society of Korea 2026
This is an Open Access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY 4.0) (https://creativecommons.org/licenses/by/4.0/) which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Supplementary Information
References
Citations
Citations to this article as recorded by 
- Next‐Generation Eco‐Omics: Integrating Microbial Function Into Predictive Ecosystem Models
Kulmani Mehar, Kamakshi Priya K, Amit Prakash Sen, Ravi Kumar Paliwal, Bhavan Kumar M., Aravindan Munusamy Kalidhas, Tapas Kumar Mohapatra, Aseel Samrat, Ravikumar Jayabal
Biotechnology and Applied Biochemistry.2026; 73(3): 1667. CrossRef - The Role of Genitourinary Microbiome in Male Cancer Etiology and Progression: Insights from Next-Generation Sequencing and Meta-Omics
Pooja Tiwary, Krishil Oswal, Ryan Varghese
Société Internationale d’Urologie Journal.2026; 7(1): 9. CrossRef - Bioinformatics in Antifungal Design: Strategies To Overcome Resistance from a Proteomic Perspective
Diego Romário-Silva, Edja Maria Melo de Brito Costa, Joanilda Paolla Raimundo Silva, Letícia Targino Campos, Vitória Marina Abrantes Batista, Camila Vital de Araújo, Sonaly Lima Albino, Arthur Gabriel Corrêa de Farias, Igor José dos Santos Nascimento, Ric
Current Fungal Infection Reports.2026;[Epub] CrossRef - 16S-Pipeline: A comprehensive web-based platform for end-to-end 16S rRNA amplicon sequencing analysis
Tatsuya Unno
Journal of Microbiology.2026; 64(5): e2603014. CrossRef - Advances in Enzymatic Production of Prebiotic Oligosaccharides from Agro-Industrial Waste: A Critical Review and Industrial Framework
Slim Smaoui
Foods.2026; 15(12): 2156. CrossRef - Activity-guided discovery of antibiotic-transforming bacteria from environmental microbiomes using D-amino acid–assisted fluorescence-activated cell sorting
Yi Liu, Kai-Li Wang, Yu-Qi Hong, Ye Yuan, Hua Wang, Yi-Qun Chen, Sheng-Song Yu, Zi-Xuan Lu, Yuan Pan, Ting-Ting Zhu
Environmental Pollution.2026; 405: 128591. CrossRef - An inferential ceiling in nanomaterial-assisted phytoremediation studies: Insights from a semi-systematic review of functional evidence in soil microbial communities
Ottavia Pinto, Marco Contin, Luca Marchiol
Applied Soil Ecology.2026; 225: 107199. CrossRef - Microbiome–metabolite signaling networks in gastrointestinal disease: systems biology, network rewiring, and precision therapeutics
Yahya A. Almutawif, Hamza M. A. Eid
Archives of Microbiology.2026;[Epub] CrossRef - Naringenin: From sustainable biosynthesis to gut microbiota-mediated bioactivation and systemic health outcomes
Shutong Liu, Tian Gong, Chenxu Zhao, Chaoqun Zhang, Yanhui Han, Hang Xiao, Yonghong Meng
Trends in Food Science & Technology.2026; 176: 105914. CrossRef - Microbiota, chronic inflammation, and health: The promise of inflammatome and inflammatomics for precision medicine and health care
Huan Zhang, Bing Jun Yang Lee, Tong Wang, Xuesong Xiang, Yafang Tan, Yanping Han, Yujing Bi, Fachao Zhi, Xin Wang, Fang He, Seppo J. Salminen, Baoli Zhu, Ruifu Yang
hLife.2025; 3(7): 307. CrossRef - Study on the Rhizosphere Soil Microbial Diversity of Five Common Orchidaceae Species in the Transitional Zone Between Warm Temperate and Subtropical Regions
Jingjing Du, Shengqian Guo, Xiaohang Li, Zhonghu Geng, Zhiliang Yuan, Xiqiang Song
Diversity.2025; 17(9): 605. CrossRef - Bioengineered Skin Microbiome: The Next Frontier in Personalized Cosmetics
Cherelle Atallah, Ayline El Abiad, Marita El Abiad, Mantoura Nakad, Jean Claude Assaf
Cosmetics.2025; 12(5): 205. CrossRef - Computational Metagenomics: State of the Art
Marco Antonio Pita-Galeana, Martin Ruhle, Lucía López-Vázquez, Guillermo de Anda-Jáuregui, Enrique Hernández-Lemus
International Journal of Molecular Sciences.2025; 26(18): 9206. CrossRef - Rotation of Corydalis yanhusuo with different crops enhances its quality and soil nutrients: a multi-dimensional analysis of rhizosphere microecology
Jia Liu, Qiang Yuan, Kejie Zhang, Xiaoxiao Sheng, Zixuan Zhu, Ning Sui, Hui Wang
BMC Plant Biology.2025;[Epub] CrossRef
 ePub Link
ePub Link Cite this Article
Cite this Article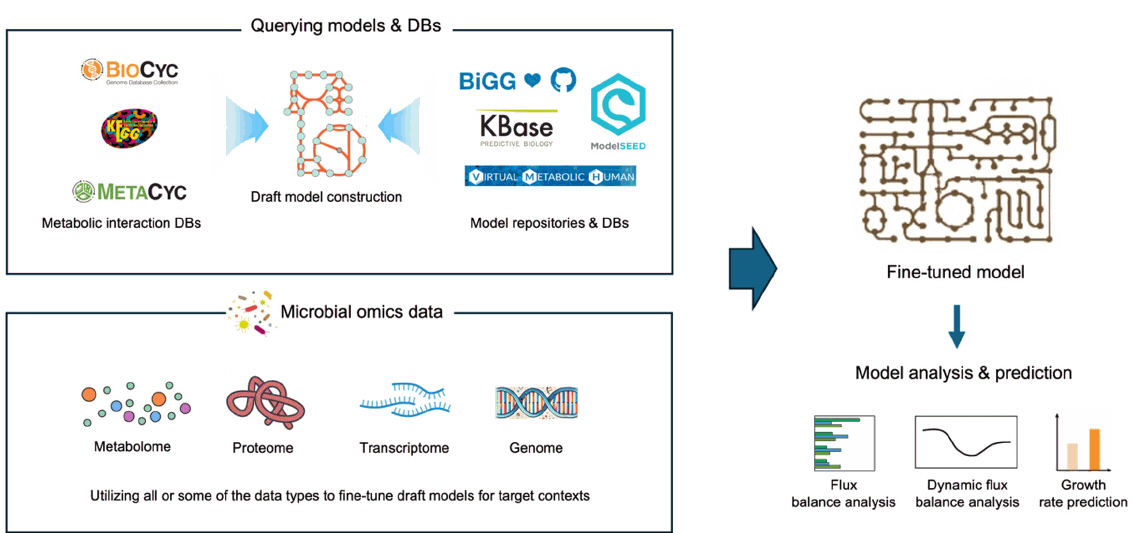
Advances in functional analysis of the microbiome: Integrating metabolic modeling, metabolite prediction, and pathway inference with Next-Generation Sequencing data
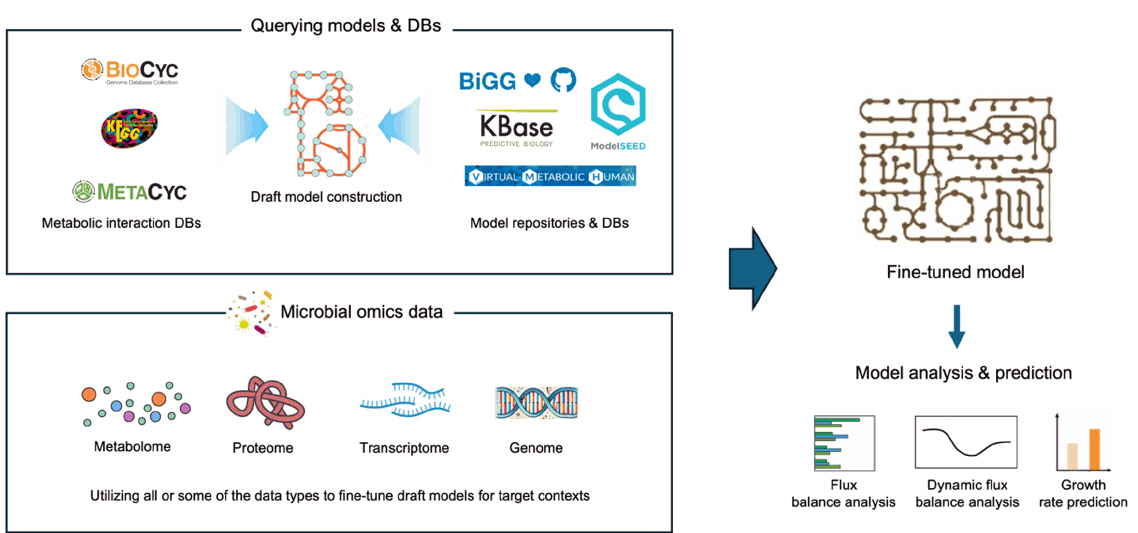
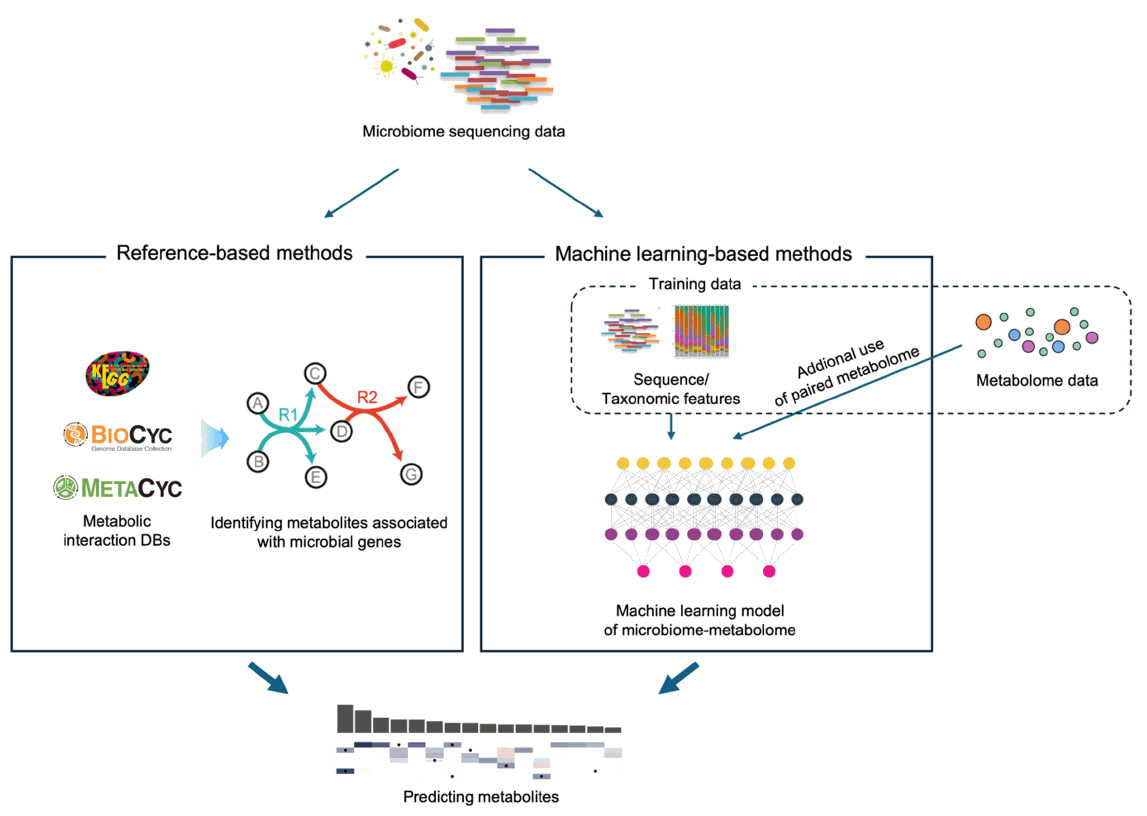
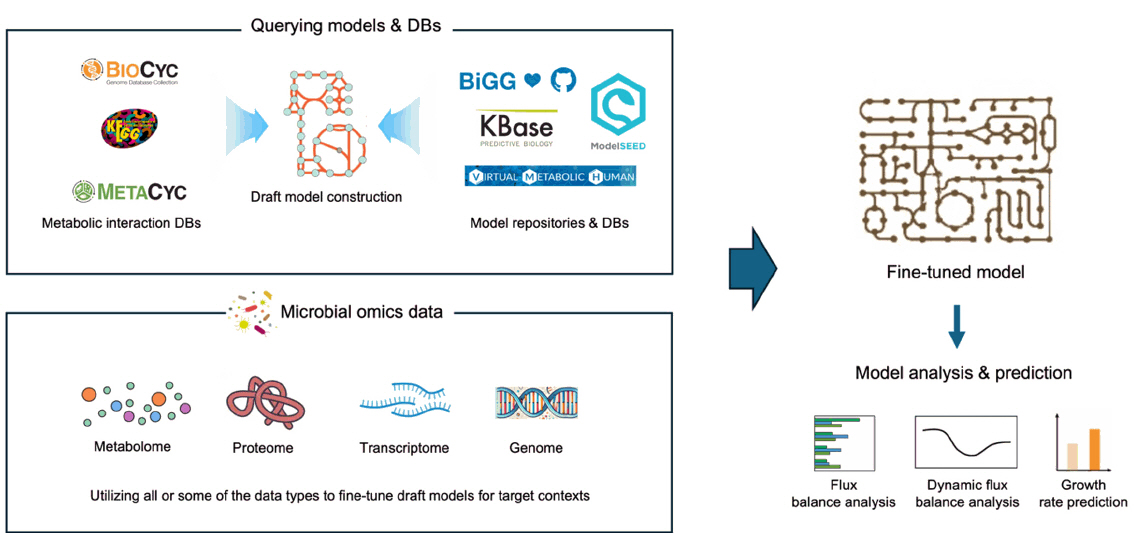
Fig. 1. The general workflow of building and using GEMs. Draft models are built based on known metabolic interactions and pre-built models, then fine-tuned based on context-specific omics profiles. Final models can be used for various purposes including the prediction of metabolite fluxes (FBA and dFBA) and the prediction of microbial growth based on metabolic activity.
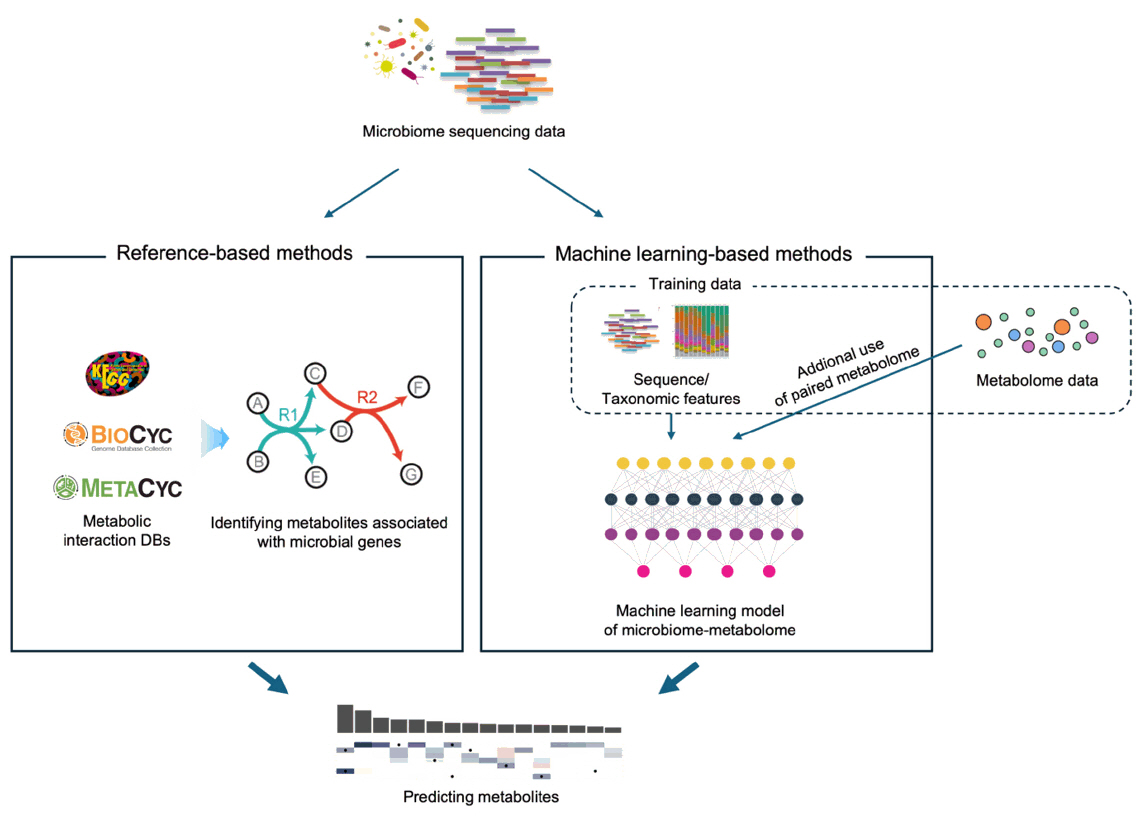
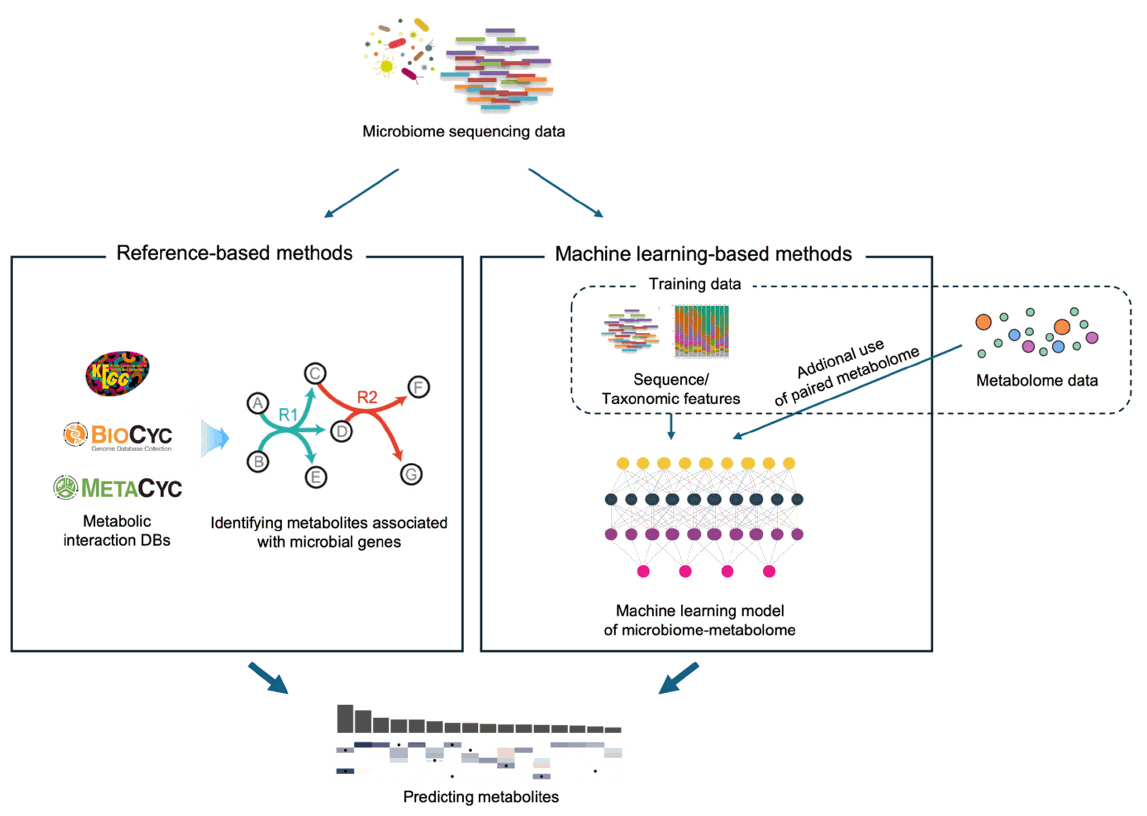
Fig. 2. Comparing the approaches of reference-based and ML-based metabolite prediction methods. Reference-based methods utilize known metabolic interactions as well as gene orthologs, while ML-based methods empirically “learn” such associations from training data.
Fig. 1.
Fig. 2.
Advances in functional analysis of the microbiome: Integrating metabolic modeling, metabolite prediction, and pathway inference with Next-Generation Sequencing data
| Name | Purpose/Functionality | Key features |
|---|---|---|
| AGORA2 ( |
Personalized and predictive modeling | Models of 7,302 microbial strains |
| Model repository | Information on 98 drugs and relevant enzymes | |
| BacArena ( |
Individual-based metabolic modeling of microbial communities | Integrates FBA with individual-based modeling |
| Modeling spatial and temporal dynamics | ||
| BiGG ( |
Repository for GEMs | 77 manually curated GEMs |
| Knowledge integration | Supporting various model formats | |
| Community collaboration | Supporting web API | |
| CarveMe ( |
Fast reconstruction of GEMs for microbial species and communities | Top-down approach using a universal model for scalable model generation |
| Automated gap-filling for improved growth phenotype predictions | ||
| COBRA ( |
Constraint-based modeling of biochemical networks | Extensive support for FBA and omics data integration |
| High-performance solvers for multi-scale and genome-scale models | ||
| COMETS ( |
Dynamic simulation of microbial community interactions | Spatially structured dFBA |
| Supports Python and MATLAB interfaces for customized simulations | ||
| DyMMM ( |
Simulating interactions and competition in microbial communities under dynamic conditions | Integrates genome-scale models for multi-species interactions |
| Predicts community dynamics under varying environmental conditions | ||
| jQMM ( |
Modeling microbial metabolism and analyzing omics data | Combines FBA and 13C metabolic flux analysis |
| Uses 13C labeling data for genome-scale model constraints | ||
| KBase ( |
Data sharing, integration, and analysis for systems biology | Diverse data integration (genomes, biochemistry) |
| Web-based interface with data provenance | ||
| MCM ( |
Modeling multi-species microbial communities with genome-based metabolic models | Statistical parameter calibration with experimental data |
| dFBA for metabolic interaction simulation | ||
| metaGEM ( |
Reconstruction of GEMs from metagenome | End-to-end pipeline for community-level metabolic interaction simulations |
| Generates personalized metabolic models from metagenome-assembled genomes (MAGs) | ||
| MetExplore ( |
Collaborative curation and exploration of metabolic networks | Data mapping for multi-omics integration |
| Sub-network extraction and interactive visualization | ||
| Microbiome Modeling Toolbox ( |
Efficient modeling and analysis of microbiome communities | Parallelized generation of personalized microbiome models |
| Visualization and statistical analysis for model comparison | ||
| MMinte ( |
Predicts metabolic interactions among microbial species in a community | Pairwise interaction analysis under different metabolic conditions |
| Modular interface with independent functionalities for flexibility | ||
| ModelSEED ( |
High-throughput generation and optimization of GEMs | Automated reconstruction pipeline from genome annotation to draft models |
| Integrates gap-filling for biomass production and growth simulation | ||
| OptCom ( |
Multi-level optimization for modeling metabolic interactions in microbial communities | Balances individual VS. community fitness criteria |
| Captures various interaction types (positive, negative) for multiple species | ||
| RAVEN ( |
Reconstruction and analysis of GEMs | Supports de novo model reconstruction using KEGG and MetaCyc databases |
| Integration with COBRA Toolbox for compatibility and bi-directional model conversion | ||
| SteadyCom ( |
Predicting microbial community composition and maintaining steady-state growth | Ensures constant community growth rate across all species |
| Supports flux variability analysis to explore metabolic flexibility | ||
| VMH ( |
Integration of models with extrinsic factors such as nutrition and disease | Extensive data coverage (Recon3D human model, 818 microbial models, disease/nutrition information) |
| Method type | Name | Data requirements | Advantages | Limitations |
|---|---|---|---|---|
| ML-based | LOCATE ( |
Paired microbiome (16S or metagenomics) and metabolomics data | Latent representation and low data requirement for training | Limited cross-dataset generalization |
| Reference-based | Mangosteen ( |
Microbiome sequencing data | Utilizes curated databases | Limited by database coverage |
| ML-based | MelonnPan ( |
Amplicon or metagenomic sequencing data, paired with metabolomic data for training | Predicts metabolomic profiles from metagenomic data | Requires training data and limited generalization |
| ML-based | MiMeNet ( |
Paired microbiome (metagenomic taxonomic/functional) and metabolome data | Improves prediction via multivariate learning | Performance depends on dataset size |
| Reference-based | MIMOSA2 ( |
Paired microbiome (16S or metagenomics) and metabolomics data | Infers mechanistic microbe-metabolite links | Limited to environments represented in reference databases |
| Name | Approach | Input data | Unique features |
|---|---|---|---|
| bioBakery ( |
Reference-based, assembly-independent profiling | Metagenomic and metatranscriptomic sequences | Integrates taxonomic, strain-level, functional, and phylogenetic profiling |
| METABOLIC ( |
High-throughput metabolic and biogeochemical profiling | Genomes from isolates, metagenome-assembled genomes, or single-cell genomes | Community-scale functional networks |
| MintTea ( |
Identification of multi-omic modules | Taxonomic, Functional, Metabolome profiles | Integration of multi-modal data and identifying predictive modules |
| PICRUSt2 ( |
Phylogenetic placement and hidden state prediction | 16S rRNA gene sequences | ASV compatibility, supports custom databases |
Table 1. Overview of GEM tools and resources
Table 2. Comparison of metabolite prediction methods
Table 3. Functional pathway analysis tools for microbiome research
Table 1.
Table 2.
Table 3.
TOP

